| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 12, Number 7, July 2021, pages 275-279
A Rare Case of Hemoglobin Bart’s Hydrops Fetalis due to Uniparental Disomy of Chromosome 16
Yin Ru Tana, b, Hak Koon Tana
aDepartment of Obstetrics and Gynaecology, Singapore General Hospital, Outram Rd, Singapore 169608, Singapore
bCorresponding Author: Yin Ru Tan, Department of Obstetrics and Gynaecology, Singapore General Hospital, Outram Rd, Singapore 169608, Singapore
Manuscript submitted March 20, 2021, accepted April 5, 2021, published online May 13, 2021
Short title: Hemoglobin Bart’s Hydrops Fetalis
doi: https://doi.org/10.14740/jmc3693
| Abstract | ▴Top |
Hemoglobin (Hb) Bart’s hydrops fetalis is the most severe form of α-thalassemia and is usually inherited in an autosomal recessive manner. We report a case of Hb Bart’s hydrops fetalis due to uniparental disomy of chromosome 16. Antenatal screening showed a low maternal mean corpuscular volume (MCV), while paternal MCV was normal. The fetus was found to have a thickened nuchal translucency during first trimester screening for Down’s syndrome. Mid-trimester fetal anomaly ultrasound scan showed fetal cardiomegaly with pericardial effusion, scalp edema, ascites and an elevated middle cerebral arterial peak systolic velocity (MCA PSV). Multiplex polymerase chain reaction (PCR) on DNA from amniocentesis showed that the fetus was homozygous for South East Asian (SEA) type 2 α-globin gene deletion. Chromosome microarray (CMA) showed two regions of absence of heterozygosity (AOH) on the terminal p and q arm of chromosome 16. The rare occurrence of Hb Bart’s hydrops fetalis caused by maternal uniparental disomy should be considered in cases of fetal hydrops even in cases where paternal MCV is normal.
Keywords: Hemoglobin Bart’s disease; Hydrops fetalis; Alpha-thalassemia; Uniparental disomy
| Introduction | ▴Top |
Thalassemia is a single gene disorder involving inherited defects in globin chain biosynthesis [1]. It can be classified into alpha (α)- and beta (β)-thalassemia. Alpha-thalassemia occurs when there is a deletion of one or more α-globin genes (located on chromosome 16), leading to reduced or absent synthesis of α-globin chains [2]. Hemoglobin (Hb) Bart’s disease involves the complete loss of all four α-globin genes (—/—), resulting in no α-globin chain production [1]. It is the most severe form of α-thalassemia, usually leading to death in utero or shortly after birth [3, 4].
Thalassemia is inherited in a simple autosomal recessive Mendelian pattern. Hb Bart’s disease is usually due to inheritance of α-thalassemia cis deletion (—/αα) from each parent [3] and couples who are both carriers of this deletion have a 25% chance of having a child affected by Hb Bart’s hydrops fetalis syndrome [4]. We present a case of Hb Bart’s hydrops fetalis syndrome due to uniparental disomy.
| Case Report | ▴Top |
Investigations
The patient was a 32-year-old gravida 2 para 0 Chinese woman with a previous history of a right tubal ectopic pregnancy and a family history of thalassemia. She had an early dating scan at 8 weeks of gestation. First trimester Down’s syndrome screening was performed at 12 + 2 weeks of gestation, with ultrasound scan showing that fetal nasal bone was present, but fetal nuchal translucency was thickened at 5 mm (Fig. 1). Adjusted risks of trisomy 21 (Down’s syndrome), trisomy 18 (Edwards’ syndrome) and trisomy 13 (Patau’s syndrome) were 1 in 30, 1 in 25, and 1 in 352, respectively. Non-invasive prenatal testing revealed a low probability of trisomy 21, 18 and 13, low probability of sex chromosome aneuploidy and no evidence of Digeorge syndrome. Antenatal blood tests done showed that the patient had an Hb level of 9.6 g/dL with low MCV of 72.9 fL. Hb electrophoresis did not reveal any abnormal Hb band and blood group and antibody screen showed that her blood group was O+ with no significant red cell antibodies. Her husband had a normal Hb (15.6 g/dL) and MCV (86.0 fL). His Hb electrophoresis did not reveal any abnormal Hb band. The patient underwent amniocentesis at 16 + 1 weeks of gestation. Amniotic fluid sent for karyotyping showed a female karyotype (46XX) with no apparent chromosomal abnormalities. Chromosome microarray (CMA) was also sent.
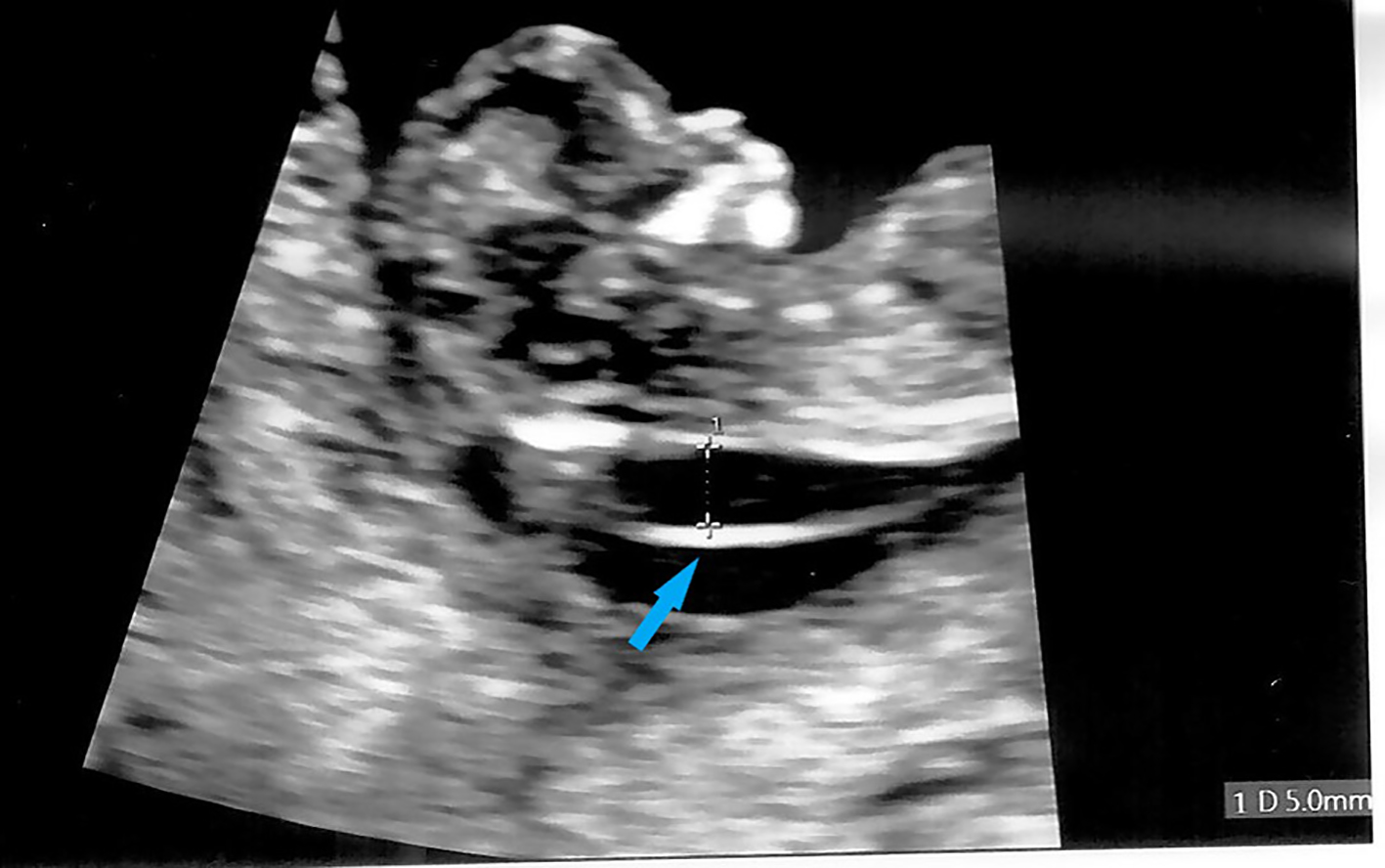 Click for large image | Figure 1. First trimester ultrasound showing fetal thickened nuchal translucency (arrow). |
Diagnosis
Mid-trimester fetal anomaly ultrasound scan at 19 weeks showed signs of hydrops fetalis. An enlarged fetal heart with pericardial effusion (Fig. 2) and suspicion of possible atrial septal defect (ASD) was noted. Scalp edema (Fig. 3) and ascites (Fig. 4) were also seen. Fetal middle cerebral arterial peak systolic velocity (MCA PSV) was also elevated at 41 cm/s (> 1.5 MoM) suggesting fetal anemia. Fetal 2D echocardiogram at 20 + 4 weeks of gestation showed moderate pericardial effusion, moderate secundum ASD and mild tricuspid regurgitation. Doppler ultrasound showed persistently elevated MSA PSV at 43 cm/s (> 1.5 MoM). Infective screening tests for cytomegalovirus, toxoplasmosis and parvovirus were negative. Multiplex polymerase chain reaction (PCR) indicated that the patient was a heterozygous carrier of the SEA type 2 α-globin gene (-SEA) deletion and her husband did not carry any α-globin gene deletion. Multiplex PCR on DNA from amniocentesis showed that the fetus was homozygous for SEA type 2 α-globin gene deletion. Maternal contamination was excluded. CMA showed two regions of absence of heterozygosity (AOH) on the terminal p and q arm of chromosome 16, respectively: 16q21q24.3 and 16p13.3p13.2, which raised the suspicion of maternal uniparental disomy.
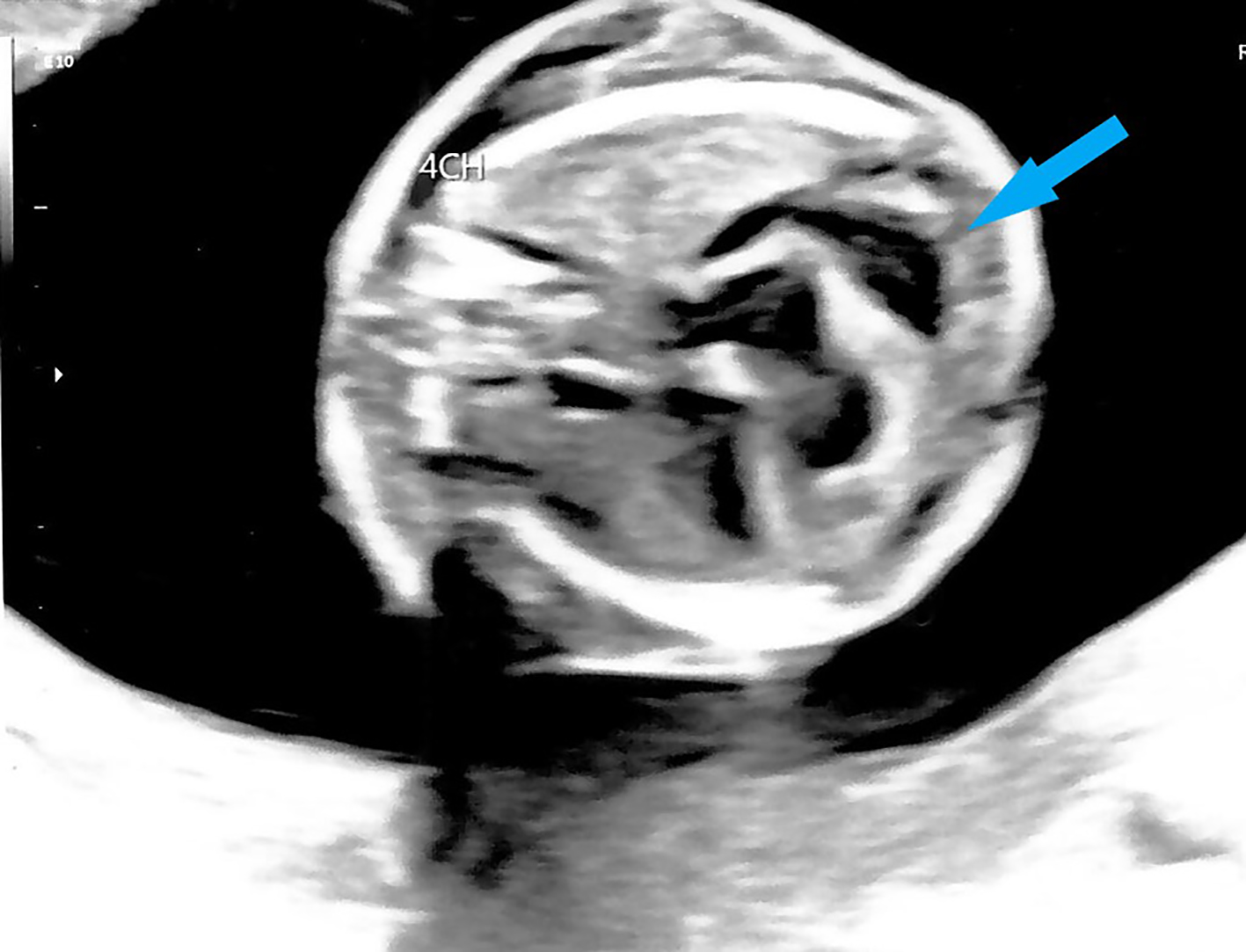 Click for large image | Figure 2. Four-chamber cardiac view on ultrasound showing fetal cardiomegaly and pericardial effusion at 19 weeks gestation (arrow). |
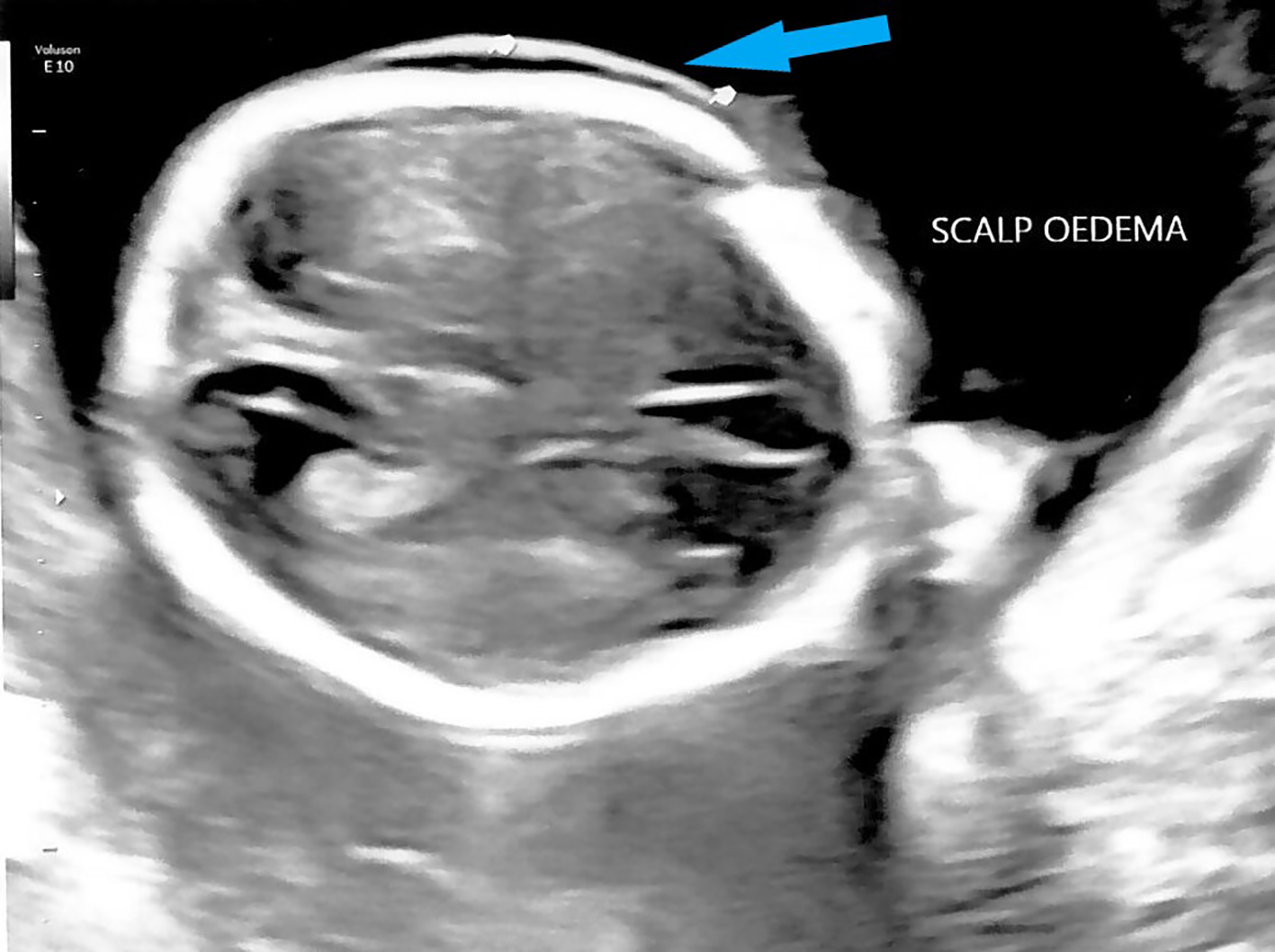 Click for large image | Figure 3. Ultrasound showing fetal scalp edema at 19 weeks gestation (arrow). |
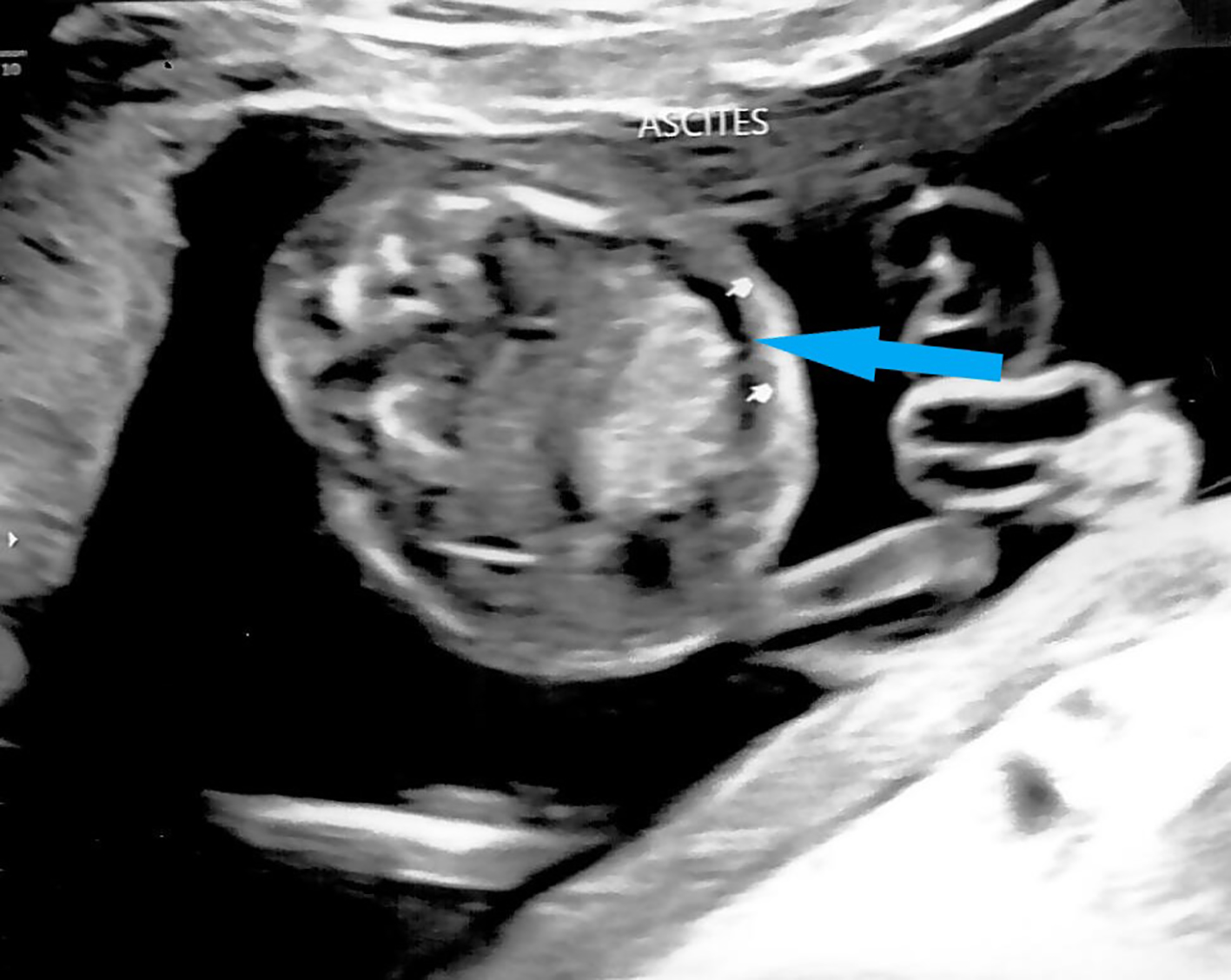 Click for large image | Figure 4. Ultrasound showing fetal ascites at 19 weeks gestation (arrow). |
Treatment and follow-up
The patient subsequently underwent termination of pregnancy at 22 weeks of gestation.
| Discussion | ▴Top |
Alpha-thalassemia is the most prevalent single gene disorder [4], with a high prevalence of 22.6% in Southeast Asia [5]. In Singapore, a multiracial Southeast Asian country, 6.4% of Chinese, 4.8% of Malays and 5.2% of Indians were found to be carriers of α-globin gene mutations [6]. Routine antenatal screening for thalassemia is conducted for all pregnant women in Singapore by measurement of maternal MCV and Hb electrophoresis. A low measurement of maternal MCV will warrant a measurement of paternal MCV. Genetic testing for α-thalassemia will also be offered. A low maternal MCV and normal paternal MCV usually do not trigger further investigation since the child should not be affected by thalassemia major syndromes if we follow the typical Mendelian autosomal recessive inheritance pattern.
We present a rare case of Hb Bart’s hydrops fetalis syndrome due to maternal uniparental disomy at chromosome 16. Uniparental disomy refers to the inheritance of a pair of chromosomes from only one parent [7]. Although maternal uniparental disomy at chromosome 16 is commonly associated with intrauterine growth restriction [8], it has been associated with unmasking of recessive conditions, such as α-thalassemia major and Fanconi anemia [9]. Several cases of maternal uniparental disomy leading to Bart’s hydrops fetalis syndrome have been reported by Kou et al, Au et al and Wattanasirichaigoon et al [10-12].
This case highlights the limitations of thalassemia screening with parental MCV. Firstly, using the threshold of MCV < 80 fL may not detect all patients who are thalassemia carriers, and there are cases of α-globin gene deletions found in patients with MCV > 80 fL [13, 14]. Moreover, similar to our case, in the cases reported by both Kou et al and Au et al, only one parent was a carrier of α-thalassemia. This suggests that a normal MCV and negative thalassemia screen in a single parent may not rule out fetal Hb Bart’s disease due to the rare occurrence of maternal uniparental disomy.
In our case, signs of fetal hydrops were noted on mid-trimester ultrasound screening. Further investigation of the cause of fetal hydrops led to the discovery that the fetus was affected by Hb Bart’s hydrops fetalis syndrome, which is the commonest cause of hydrops fetalis in Southeast Asia [15, 16]. The fetus was found to be homozygous for SEA type 2 α-globin gene deletion, which is the main cause of the Hb Bart’s hydrops fetalis in Asia [17]. This highlights the importance of a mid-trimester screening ultrasound even if only one parent is a thalassemia carrier. Although not available in all Asian countries, a mid-trimester ultrasound screening for fetal anomalies is performed for all antenatal patients in Singapore from 18 to 20 + 6 weeks as recommended by the NICE guidelines [18].
In conclusion, uniparental disomy of chromosome 16 is a rare but possible cause of Hb Bart’s hydrops fetalis and should be considered in cases with sonographic features of fetal hydrops, even in cases where only one parent is an α-thalassemia carrier.
Acknowledgments
The authors would like to acknowledge the valuable input and support from the doctors and sonographers from the Department of Obstetrics and Gynaecology, Singapore General Hospital, as well as the doctors from the Genetics Service, KK Women’s and Children’s Hospital.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Written informed consent was obtained from the patient.
Author Contributions
All authors were involved in conception and editing of the manuscript. THK was involved in the clinical care of the patient.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Giardina PJ, Rivella S. Chapter 38 - Thalassaemia Syndromes. In: Hoffman R. Hematology: basic principles and practice 6th ed. 2013. p. 505-535.e13. Elsevier.
- Muncie HL Jr, Campbell J. Alpha and beta thalassaemia. Am Fam Physician. 2009;80(4):339-344.
- Cappellini MD, Cohen A, Eleftheriou A, Piga A, Porter J, Taher A. Guidelines for the Clinical Management of Thalassaemia, 2nd Revised Ed. Nicosia (CY): Thalassaemia International Federation. 2008.
- Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2010;5:13.
doi pubmed - Goh LPW, Chong ETJ, Lee PC. Prevalence of Alpha(α)-Thalassaemia in Southeast Asia (2010-2020): a meta-analysis involving 83,674 subjects. Int J Environ Res Public Health. 2020;17(20):7354.
doi pubmed - Kham SK, Quah TC, Loong AM, Tan PL, Fraser A, Chong SS, Yeoh AE. A molecular epidemiologic study of thalassemia using newborns' cord blood in a multiracial Asian population in Singapore: results and recommendations for a population screening program. J Pediatr Hematol Oncol. 2004;26(12):817-819.
- Lapunzina P, Monk D. The consequences of uniparental disomy and copy number neutral loss-of-heterozygosity during human development and cancer. Biol Cell. 2011;103(7):303-317.
doi pubmed - Eggermann T, Curtis M, Zerres K, Hughes HE. Maternal uniparental disomy 16 and genetic counseling: new case and survey of published cases. Genet Couns. 2004;15(2):183-190.
- Donovan FX, Kimble DC, Kim Y, Lach FP, Harper U, Kamat A, Jones M, et al. Paternal or Maternal Uniparental Disomy of Chromosome 16 Resulting in Homozygosity of a Mutant Allele Causes Fanconi Anemia. Hum Mutat. 2016;37(5):465-468.
doi pubmed - Kou KO, Lee H, Lau B, Wong WS, Kan A, Tang M, Lau ET, et al. Two unusual cases of haemoglobin Bart's hydrops fetalis due to uniparental disomy or non-paternity. Fetal Diagn Ther. 2014;35(4):306-308.
doi pubmed - Au PK, Kan AS, Tang MH, Leung KY, Chan KY, Tang TW, Lau ET. A Fetus with Hb Bart's disease due to maternal uniparental Disomy for chromosome 16. Hemoglobin. 2016;40(1):66-69.
doi pubmed - Wattanasirichaigoon D, Promsonthi P, Chuansumrit A, Leopairut J, Yanatatsaneejit P, Rattanatanyong P, Munkongdee T, et al. Maternal uniparental disomy of chromosome 16 resulting in hemoglobin Bart's hydrops fetalis. Clin Genet. 2008;74(3):284-287.
doi pubmed - Sirichotiyakul S, Maneerat J, Sa-nguansermsri T, Dhananjayanonda P, Tongsong T. Sensitivity and specificity of mean corpuscular volume testing for screening for alpha-thalassemia-1 and beta-thalassemia traits. J Obstet Gynaecol Res. 2005;31(3):198-201.
doi pubmed - Chan LC, Ma SK, Chan AY, Ha SY, Waye JS, Lau YL, Chui DH. Should we screen for globin gene mutations in blood samples with mean corpuscular volume (MCV) greater than 80 fL in areas with a high prevalence of thalassaemia? J Clin Pathol. 2001;54(4):317-320.
doi pubmed - Tan SL, Tseng AM, Thong PW. Bart's hydrops fetalis - clinical presentation and management - an analysis of 25 cases. Aust N Z J Obstet Gynaecol. 1989;29(3 Pt 1):233-237.
doi pubmed - Liang ST, Wong VC, So WW, Ma HK, Chan V, Todd D. Homozygous alpha-thalassaemia: clinical presentation, diagnosis and management. A review of 46 cases. Br J Obstet Gynaecol. 1985;92(7):680-684.
doi pubmed - He S, Li J, Huang P, Zhang S, Lin L, Zuo Y, Tian X, et al. Characterization of Hb Bart's hydrops fetalis caused by - -(SEA) and a large novel alpha(0)-thalassemia deletion. Hemoglobin. 2018;42(1):61-64.
doi pubmed - National Institute for Health and Care Excellence. Antenatal care for uncomplicated pregnancies (NICE Guideline No. 62). Available at: https://www.nice.org.uk/guidance/cg62. 2020. Accessed March 9, 2021.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.