

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 4, Number 4, April 2013, pages 254-258
Effect of Insulin Sensitizers in Three Patients With ACTH-Dependent Hypercortisolism
Alan Sacerdotea, e, Gul Bahtiyara, MaryAnn Banerjib, Geeti Mahajanb, Marie-Alex Michel Vincentc, Richard Vincentd
aDivision of Endocrinology, Woodhull Medical Center, 760 Broadway, Brooklyn, NY 11206, USA
bDivision of Endocrinology, SUNY Downstate Medical Center, 450 Clarkson Avenue, Brooklyn, NY 11203, USA
cDivision of Nephrology, Woodhull Medical Center, 760 Broadway, Brooklyn, NY 11206, USA
dCollege of Liberal Arts, Hofstra University, 516 Harvard Avenue, Baldwin, NY 11510, USA
eCorresponding author: Alan Sacerdote, Division of Endocrinology, Woodhull Medical Center, 760 Broadway, Brooklyn, NY 11206, USA
Manuscript accepted for publication January 22, 2013
Short title: Insulin Sensitizers and Hypercortisolism
doi: https://doi.org/10.4021/jmc1061w
| Abstract | ▴Top |
Three women 40, 67, and 50 years old had ACTH-dependent hypercortisolism (ADH), 2/3s with diabetes (DM). Metformin alone, or in combination with the PPAR-γ agonist, pioglitazone, normalized UFC in our patients and lowered plasma ACTH. With insulin sensitizer (IS) treatment, all patients experienced a decrease in UFC from baseline ≥ 90 %. Plasma ACTH also fell from baseline 15-20 % in all patients. Our findings suggest that ADH may be an expression of insulin resistance or that insulin resistance exerts a permissive effect on the expression of ADH in some patients. Insulin sensitizers (IS) may be an alternative treatment for some ADH patients.
Keywords: ACTH-dependent; Hypercortisolism; Insulin resistance; Insulin sensitizer; Metformin; Thiazolidinediones
| Introduction | ▴Top |
It’s been reported that about 3% of Type 2 diabetics have CD [1]. Many Cushing’s disease (CD) patients have peroxisome proliferator activator receptor - gamma (PPAR-γ) in their adenomas. Ambrosi et al [2] reported that some patients with CD respond to the insulin sensitizer, rosiglitazone, with a reduction in both urinary free cortisol and plasma ACTH. Suri and Weiss [3] reported, in a small series, that pioglitazone monotherapy was ineffective in ameliorating Cushing’s disease. These cases we present are the first report of amelioration of ADH with metformin either as monotherapy or in combination with pioglitazone. Our findings, if corroborated in larger, randomized controlled trials, could explain why the reported prevalence of subclinical hypercortisolism (SH) in Type 2 diabetics has recently declined and could offer an alternative medical treatment for some ADH patients. If expanded to larger studies, our findings may also explain why SH is more commonly encountered in in-patient series (in whom insulin sensitizers are typically held on hospital admission) than in ambulatory series in which these agents are generally continued. Our findings may also help to explain why the preponderance of SH reported in Type 2 diabetics is of autonomous adrenal rather than ACTH dependent origin.
| Case Report | ▴Top |
We reviewed the records of three women (aged 40, 50, and 67 years) from our endocrine clinic who were diagnosed with ADH. Our patients had clinical features of type 2 DM, as well as facial mooning, plethora, hirsutism, acanthosis, hypertension, menstrual irregularity, post-menopausal osteoporosis, depression, and abdominal striae. Not every patient had all of these features.
ADH was documented in all 3 patients by a combination of an elevated 24-hour urinary free cortisol (UFC), a non-suppressed plasma ACTH, a positive low dose dexamethasone suppression test, a normal high dose dexamethasone test, and in one case, pituitary imaging consistent with adenoma. Plasma ACTH was measured by chemiluminesence, urinary free cortisol (UFC) by high performance liquid chromatography (HPLC), and serum cortisol by chemiluminesence. Adequacy of 24 hour urine collection was assessed by creatinine and volume. Pituitary imaging was by gadolinium-enhanced MRI. Patient 3 had a prominently sized pituitary but no well defined adenoma. Patient 2 had a 3 mm adenoma as observed on the MRI.
Prior to investigation, differential diagnosis included: 1) polycystic ovarian syndrome; 2) post-menopausal ovarian hyperandrogenism (hyperthecosis); 3) non-classical congenital adrenal hyperplasia; and 4) glucocorticoid resistance syndrome.
Each of the patients gave their informed consent for the use of insulin sensitizers (IS), after discussing the potential risks and benefits of such treatment as well as alternative treatment as well as for the follow-up laboratory determinations and imaging. In the 2 patients who are diabetic, it was explained that insulin sensitizers were part of standard treatment for Type 2 diabetes, but not for ADH. It was explained to the second and third patients that the first patient had experienced biochemical and clinical improvement with insulin sensitizers and we hoped to see if they would realize similar benefits.
If treatment was initiated with metformin, it was started at 500 mg/day and titrated at each visit until both UFC and glucose were controlled, a total daily dose of 2,000 mg/day was reached, or the maximum tolerated daily dose was reached. In one patient UFC and glucose control had not been achieved, hence, pioglitazone starting at 15 mg/day was added. Patient 1 was on metformin 500 mg bid. Patient 2 was on metformin 1,000 mg bid and pioglitazone 15 mg daily. Patient 3 was on metformin 1,000 mg bid. All patients tolerated their treatments well.
Baseline clinical characteristics of patients along with the on-therapy observations are listed in Table 1. All patients initially had elevated UFC levels and elevated or inappropriately non-suppressed plasma ACTH. After treatment with insulin sensitizers, all patients decreased their UFC levels and their Plasma ACTH levels (Fig. 1-3). In patient 1 UFC fell from 193.2 to 19.6 µg/day - an 89.9 % decrease. Plasma ACTH fell from 47 to 40 pg/mL - a 14.9 % decrease from baseline. Patient 2’s UFC fell from 110 to 2.8 µg/day - a 97.5 % decrease. Her plasma ACTH fell from 35 to 28 pg/mL - a 20 % decrease. Patient 3’s UFC fell from 376.4 to 7.8 µg/day - a 97.9 % decrease. Plasma ACTH went from 34 to 27 pg/mL - a 20.6 % decrease. Also observed in patient 3 was a negative low dose dexamethasone suppression test on insulin sensitizers.
 Click to view | Table 1. Patient Characteristics |
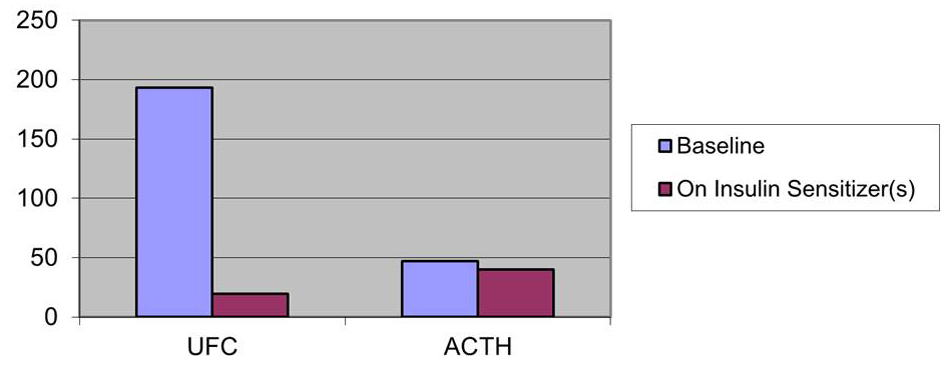 Click for large image | Figure 1. Effect of insulin sensitizers on UFC (µg/day) and ACTH (pg/mL) patient 1. |
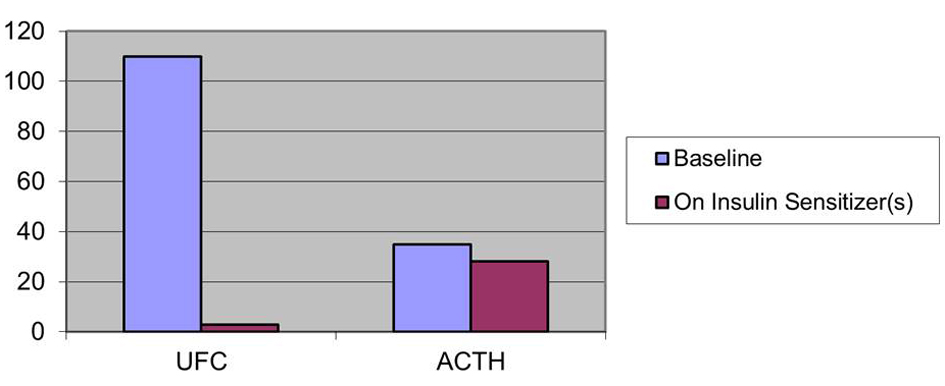 Click for large image | Figure 2. Effect on insulin sensitizers on UFC (µg/day) and ACTH (pg/mL) patient 2. |
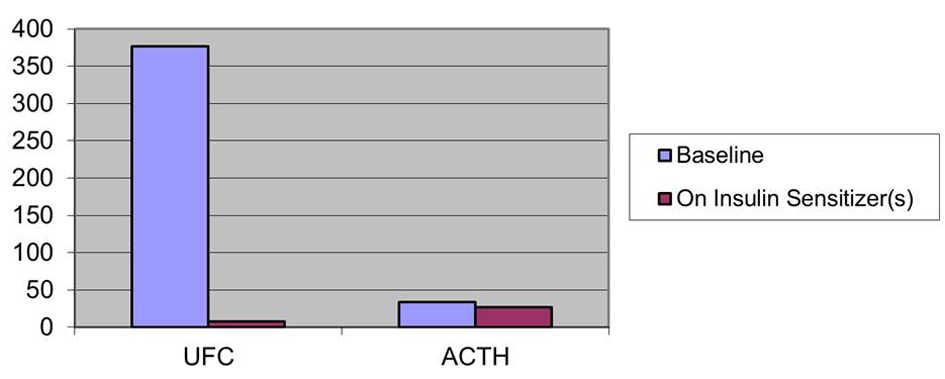 Click for large image | Figure 3. Effect on insulin sensitizers on UFC (µg/day) and ACTH (pg/mL) patient 3. |
No adverse effects on body weight, blood glucose, hemoglobin A1C, blood pressure, or fluid retention were observed as a result of the IS therapy. None of the patients experienced symptoms of adrenal insufficiency on treatment, despite quite low UFC. On insulin sensitizer treatment our patients also experienced amelioration of facial mooning, plethora, striae, hirsutism, hypertension, and hyperglycemia.
In general, the on-therapy plasma ACTH and UFC were checked on the following clinic visit (2 - 3 months). Effects of the insulin sensitizers were observable as early as 2 months after treatment initiation in 1 patient and approximately 8 - 12 months after treatment initiation in 2 patients. As of November, 2012 all 3 patients continue to have normal UFC and plasma ACTH levels.
| Discussion | ▴Top |
Although it has been previously reported that patients with CD have PPAR-γ in their adenomas [4], a recent study found that the anti-proliferative effect of the glitazones on anterior pituitary cells is independent of the PPAR-γ [5]. The exact mechanism by which the glitazones may reduce the plasma level of ACTH and the UFC is not completely understood. It has been proposed that PPAR-γ agonist binding to its nuclear receptor inhibits the synthesis and secretion of ACTH in the adenoma, however, this does not adequately explain why rosiglitazone monotherapy is effective in some patients, while pioglitazone monotherapy is not [2, 3]; nor does it explain why, in patients who do respond to rosiglitazone monotherapy, the reduction in UFC is of a greater magnitude than the reduction in plasma ACTH. It also does not explain why some patients who have PPAR-γ positive adenomas have no clinical/biochemical response to rosiglitazone monotherapy. PPAR-γ has also been reported to be present in the adrenal cortex [6], however, this does not explain the response to metformin monotherapy seen in some of our patients or the enhanced response to the glitazones seen in others with the addition of metformin.
While it is well known that ADH is an important cause of insulin resistance, our data suggest that the converse may be true in some patients-that insulin resistance may be a cause as well as a result of ADH. There appears to be considerable cross-talk between insulin and the adrenal cortex. We have reported other adrenal disorders which are ameliorated with either metformin or glitazones [7-9], including non-classical adrenal hyperplasia, drug-induced adrenal hyperandrogenism, primary hyperaldosteronism, and classical 21-hydroxylase deficiency. Insulin resistance has been reported in both non-classical and classical 21-hydroxylase deficiency [10-15]. Non-classical aldosterone synthase deficiency is ameliorated by exercise and weight loss which improve insulin sensitivity, while non-classical 11-hydroxylase deficiency may be ameliorated by Roux-en-y gastric bypass, which is also an insulin sensitizing treatment [16].
Possible mechanisms of action for metformin in the treatment of ADH may be extrapolated from the observed anti-neoplastic, anti-hyperplastic effects of this drug reported by Buzzai et al [17] and reviewed recently by Ben Sahra et al [18]. It has been shown that metformin selectively inhibits p53-deficient tumor cell growth in isogenic HCT 116 colon cancer cell line xenografts, an action that is associated with activation of AMP-activated protein kinase (AMPK) and increased tumor cell apoptosis. Metformin suppressed mitochondrial electron transport, which was compensated for by an increased rate of anaerobic glycolysis in p53 replete, but not in p53-null tumor cells.
Epidemiologic studies show a decreased cancer incidence in metformin-treated patients. Metformin decreases insulin resistance, thereby indirectly reducing serum insulin concentration, which should be beneficial because insulin promotes the growth of many types of cancer cells, both directly by binding to the IGF-1 receptor and indirectly by decreasing the concentration of IGF-binding protein, thus increasing serum free IGF-1. Metformin activates the AMPK pathway, a major sensor of the energy status of cells, which, in turn downregulates the mTOR pathway, which has been implicated in the control of protein synthesis and cellular proliferation and which is activated by mitogen-responsive pathways, for example, RAS/ERK and PI3K/AKT. Metformin has also been shown to decrease the cross-talk between insulin and the G-protein coupled receptor which serves as a receptor for a number of growth factors, including IGF-2, EGF-1, and PDGF. Since ADH involves cellular proliferation, both at the level of the pituitary, resulting in corticotroph adenoma or hyperplasia, as well as in the adrenal cortex, resulting in ACTH-dependent hyperplasia, it is plausible that metformin may inhibit cellular proliferation at both sites.
Since metformin has its most dramatic effect on p53 null tumor cells it is worth noting that 50% of corticotroph adenomas have been reported to harbour p53 mutations [19, 20].
Recently there has been some controversy concerning the prevalence of SH in patients with Type 2 DM [21, 22]. The older reports tend to report a higher prevalence of SH in Type 2 DM cohorts. Terzolo et al astutely point out that earlier series disproportionately screened hospital in-patients-a sicker, more stressed population than the routine visit ambulatory Type 2 DM patients that they screened. Severely stressed patients tend to have more transient disruption of the hypothalamic-pituitary-adrenal axis than ambulatory patients [22]. Unlike the series reported by Mullan et al [21] they appropriately excluded patients taking thiazolidinediones, because, as they noted, this class of drugs suppresses the hypothalamic-pituitary-adrenal axis. When patients are admitted to the hospital, not only are they sicker and more stressed, but because of that fact, oral agents such as metformin and thiazolidinediones are usually held and replaced with insulin, potentially unmasking SH which was actually being treated with the insulin sensitizers. Earlier studies on the prevalence of SH in Type 2 DM patients are likely to have included a lower percentage of patients on insulin sensitizers, even in ambulatory patients, simply because they were not yet in such general use. Terzolo et al also note that the preponderance of SH patients in their series and in other series are those with autonomous adrenal disease (adenoma or nodular hyperplasia), rather than ADH. It is tempting to speculate that insulin sensitizer treatment may select out those patients with autonomous adrenal disease, while actually treating and thereby masking, a number of patients with ADH, much like antibiotics may select out bacterial strains resistant to the antibiotic. Our findings suggest that if screening for CD is done in the Type 2 DM-pre-diabetic population it should be accomplished before initiating treatment with insulin sensitizers.
Limitations of this paper include: small patient number, absence of male patients, and relatively short period of follow-up. Strengths of this study include: consistent direction of response of UFC and plasma ACTH to insulin sensitizer therapy.
Grant Support
None.
Disclosures
The authors have no conflicts of interest to disclose.
| References | ▴Top |
- Catargi B, Rigalleau V, Poussin A, Ronci-Chaix N, Bex V, Vergnot V, Gin H, et al. Occult Cushing's syndrome in type-2 diabetes. J Clin Endocrinol Metab. 2003;88(12):5808-5813.
doi pubmed - Ambrosi B, Dall'Asta C, Cannavo S, Libe R, Vigo T, Epaminonda P, Chiodini I, et al. Effects of chronic administration of PPAR-gamma ligand rosiglitazone in Cushing's disease. Eur J Endocrinol. 2004;151(2):173-178.
doi pubmed - Suri D, Weiss RE. Effect of pioglitazone on adrenocorticotropic hormone and cortisol secretion in Cushing's disease. J Clin Endocrinol Metab. 2005;90(3):1340-1346.
doi pubmed - Heaney AP, Fernando M, Yong WH, Melmed S. Functional PPAR-gamma receptor is a novel therapeutic target for ACTH-secreting pituitary adenomas. Nat Med. 2002;8(11):1281-1287.
doi pubmed - Emery MN, Leontiou C, Bonner SE, Merulli C, Nanzer AM, Musat M, Galloway M, et al. PPAR-gamma expression in pituitary tumours and the functional activity of the glitazones: evidence that any anti-proliferative effect of the glitazones is independent of the PPAR-gamma receptor. Clin Endocrinol (Oxf). 2006;65(3):389-395.
doi pubmed - Betz MJ, Shapiro I, Fassnacht M, Hahner S, Reincke M, Beuschlein F. Peroxisome proliferator-activated receptor-gamma agonists suppress adrenocortical tumor cell proliferation and induce differentiation. J Clin Endocrinol Metab. 2005;90(7):3886-3896.
doi pubmed - Bahtiyar G, Weiss K, Sacerdote AS. Novel endocrine disrupter effects of classic and atypical antipsychotic agents and divalproex: induction of adrenal hyperandrogenism, reversible with metformin or rosiglitazone. Endocr Pract. 2007;13(6):601-608.
pubmed - Mapas-Dimaya AC, Agdere L, Bahtiyar G, Mejia JO, Sacerdote AS. Metformin-responsive classic salt-losing congenital adrenal hyperplasia due to 21-hydroxylase deficiency: a case report. Endocr Pract. 2008;14(7):889-891.
pubmed - Sacerdote A, L'Eplattanier ML, Toossi A, Bahtiyar G, Girgis E. Metformin-suppressible primary hyperaldosteronism: Another facet of the metabolic syndrome? Pract Diabetol 2006;25(4):37-40.
- Saygili F, Oge A, Yilmaz C. Hyperinsulinemia and insulin insensitivity in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency: the relationship between serum leptin levels and chronic hyperinsulinemia. Horm Res. 2005;63(6):270-274.
doi pubmed - Speiser PW, Serrat J, New MI, Gertner JM. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. 1992;75(6):1421-1424.
doi pubmed - Singer F, Bhargava G, Poretsky L. Persistent insulin resistance after normalization of androgen levels in a woman with congenital adrenal hyperplasia. A case report. J Reprod Med. 1989;34(11):921-922.
pubmed - Ambroziak U, Bednarczuk T, Ginalska-Malinowska M, Malunowicz EM, Grzechocinska B, Kaminski P, Bablok L, et al. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency - management in adults. Endokrynol Pol. 2010;61(1):142-155.
pubmed - Charmandari E, Chrousos GP. Metabolic syndrome manifestations in classic congenital adrenal hyperplasia: do they predispose to atherosclerotic cardiovascular disease and secondary polycystic ovary syndrome? Ann N Y Acad Sci. 2006;1083:37-53.
doi pubmed - Atabek ME, Kurtoglu S, Keskin M. Female pseudohermaphroditism due to classical 21-hydroxylase deficiency and insulin resistance in a girl with Turner syndrome. Turk J Pediatr. 2005;47(2):176-179.
pubmed - Gul Bahtiyar and Alan Sacerdote (2011). Management Approaches to Congenital Adrenal Hyperplasia in Adolescents and Adults; Latest Therapeutic Developments, Amenorrhea, Amar Chatterjee (Ed.), ISBN: 978-953-307-988-2, InTech, Available from: http://www.intechopen.com/articles/show/title/management-approaches-to-congenital-adrenal-hyperplasia-in-adolescents-and-adults-latest-therapeutic
- Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67(14):6745-6752.
doi pubmed - Ben Sahra I, Le Marchand-Brustel Y, Tanti JF, Bost F. Metformin in cancer therapy: a new perspective for an old antidiabetic drug? Mol Cancer Ther. 2010;9(5):1092-1099.
doi pubmed - Buckley N, Bates AS, Broome JC, Strange RC, Perrett CW, Burke CW, Clayton RN. P53 protein accumulates in Cushings adenomas and invasive non-functional adenomas. J Clin Endocrinol Metab. 1995;80(2):4 p following 692.
- Kawashima ST, Usui T, Sano T, Iogawa H, Hagiwara H, Tamanaha T, Tagami T, et al. P53 gene mutation in an atypical corticotroph adenoma with Cushing's disease. Clin Endocrinol (Oxf). 2009;70(4):656-657.
doi pubmed - Mullan K, Black N, Thiraviaraj A, Bell PM, Burgess C, Hunter SJ, McCance DR, et al. Is there value in routine screening for Cushing's syndrome in patients with diabetes? J Clin Endocrinol Metab. 2010;95(5):2262-2265.
doi pubmed - Terzolo M, Reimondo G, Chiodini I, Castello R, Giordano R, Ciccarelli E, Limone P, et al. Screening of Cushing's syndrome in outpatients with type 2 diabetes: results of a prospective multicentric study in Italy. J Clin Endocrinol Metab. 2012;97(10):3467-3475.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.