

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 5, Number 8, August 2014, pages 455-458
Pyoderma Gangrenosum in a Patient With Systemic Lupus Erythematosus: Case Report
Amad Hania
Department of general medicine, Senior House Officer, St James Hospital, Dublin, Ireland
Manuscript accepted for publication July 1, 2014
Short title: PG With SLE
doi: https://doi.org/10.14740/jmc1845w
| Abstract | ▴Top |
Pyoderma gangrenosum (PG) is a rare and unusual manifestation of systemic lupus erythematosus (SLE). It deserves special mention whenever a case of PG is seen in any patient population, due to its rarity. It has an unusual association with SLE, with few documented cases. A case of a 53-year-old male patient, who had an unusual first presentation of SLE in 1995, with mainly hematological and renal manifestations of SLE and with a lack of any dermatological or musculoskeletal features until 2012 is presented here. The patient developed two ulcerating lesions on right leg and was diagnosed with PG following biopsy, which was successfully treated with corticosteroids and appropriate wound management.
Keywords: Pyoderma gangrenosum; Systemic lupus erythematosus; Neutrophilic dermatosis; Antiphospholipid syndrome
| Introduction | ▴Top |
Pyoderma gangrenosum (PG) is a rare inflammatory and ulcerative disorder of the skin, which is characterized by neutrophilic infiltrates on biopsy. The most common presentation of PG is an inflammatory papule or pustule that progresses to a painful ulcer with a violaceous undermined border and a purulent base. More than 50% of patients with PG have an associated systemic disease, most commonly inflammatory bowel disease (14-34%), arthropathies (11-25%) and hematologic disease or hematologic malignancy (20%) [1]. Systemic lupus erythematosus (SLE) is not a commonly recognized associate of PG. PG is diagnosed clinically, based on its typical clinical presentation. Diagnosis is helped by excluding diseases which share a similar clinical picture such as vascular ulcers, pressure sores, trauma and infection, as the histopathologic findings of PG are nonspecific. A case of a 53-year-old Caucasian male with known SLE, presenting with PG is described in this report.
| Case Report | ▴Top |
A 53-year-old Caucasian male patient was diagnosed with SLE in 1995, following an episode of right upper quadrant pain. Patient was found to have low hemoglobin of 9 g/dL (normal 3 - 18 g/dL), a raised bilirubin 40 µmol/L (normal 3 - 17 µmol/L), low platelets (< 100 × 109/L), raised APTT 50 s (normal 28 - 38 s), raised creatinine 200 µmol (normal 60 - 110 µmol), and proteinuria. Further investigations showed patient to have positive direct Coombs test, a strongly positive anti-double stranded DNA of 150 IU/mL normal (< 30.0 IU/mL) and a positive antiphospholipid and anticardiolipin antibody. Patient was subsequently diagnosed with SLE, autoimmune hemolytic anemia, antiphospholipid syndrome and lupus nephritis. Patient had previously been well, with no previous hospital admissions, and was not on any regular medications. Patient was treated with aspirin and plaquenil and recovered well following a cholecystectomy for cholecystitis. Patient subsequently emigrated to Ireland from UK a few years following the diagnosis. Patient was started on regular low dose steroids (prednisolone 7.5 mg) following flare up of autoimmune hemolytic anemia and warfarin following a deep vein thrombosis in 2002.
On December 9, 2012, patient noticed a well-defined irregular papular lesion on right mid shin (Fig. 1). The patient attended his GP 2 days later. The lesion failed to resolve with oral antibiotics for a presumed infective etiology, and the lesion progressed to a painful 2 × 2 cm well-defined ulcerating lesion with surrounding erethyma in the following week and subsequently referred to Dermatology in St James Hospital. A second 1 × 1 cm ulcerating lesion appeared just below the first lesion (Fig. 2, 3) a few days later. Both lesions had well-defined purplish undermined borders and a necrotic base with surrounding eretheyma, and there was marked tenderness on palpation. There was no history of trauma, no features of systemic illness such as cough, shortness of breath, rigors, abdominal pain, diarrhea, joint pain, or any other vasculitic rash. On examination, patient was afebrile and all vital signs were stable. Peripheral pulses were normal on palpation and sensations over both lower limbs were normal. Investigations revealed a hemoglobin of 10 g/dL (normal 3 - 18 g/dL), white blood cell count 12 × 109/L (normal 4 × 109/L - 11 × 109/L), raised CRP 150 mg/L (normal 0 - 10 mg/L) and a fasting glucose of 5 mmol/L (normal 3.5 - 5 mmol/L). A plain film of right leg was normal. A Doppler ultrasound of right leg showed no evidence of venous obstruction. An arterial sonography scan showed no evidence of arterial insufficiency. A further immunology screen was carried out and patient remained to have a positive anti-double stranded DNA. An antineutrophilic cytoplasmic antibody (c ANCA) and (p ANCA) was negative. The patient was also found to have no evidence of inflammation in OGD and colonoscopy.
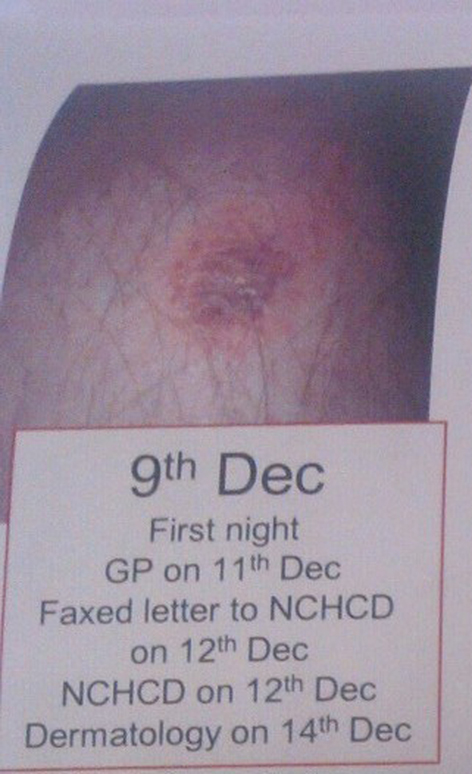 Click for large image | Figure 1. Well-defined tender papular lesion on right shin, on day 1. |
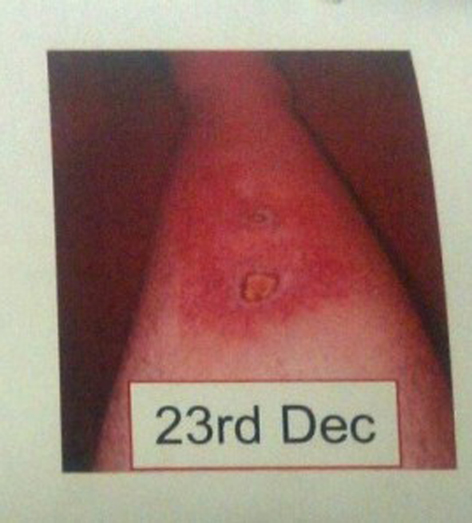 Click for large image | Figure 2. Progression of initial popular lesion to a 2 × 2 cm painful ulcerating lesion with surrounding erethyma, and a second 1 × 1 cm ulcer just below. Both ulcers have violaceous border and exudative base, on day 15. |
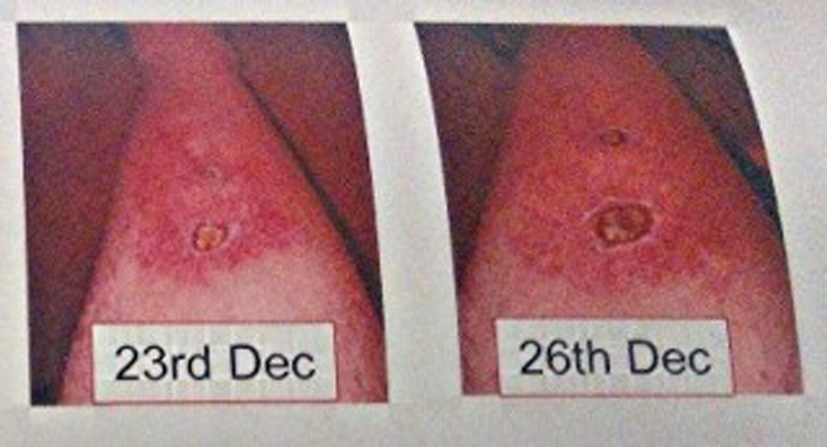 Click for large image | Figure 3. Progression of ulcers, with an increasing size of the proximal ulcer from 2 × 2 cm to 3 × 3 cm and with increased surrounding eretheyma, on day 15 and day 18 respectively. |
A clinical diagnosis of PG was made based on features of rapid onset and progression, resistance to treatment with antibiotics, and characteristic of lesion (ulcerating, well-defined purple undermined borders), biopsy that showed neutrophilic infiltrates with patches of necrosis of both dermis and epidermis. Patient was treated with systemic steroids (prednisolone 60 mg) tapering over 6 months, an increase in regular dose, and appropriate wound care measures and dressings to prevent infection and promote wound healing. The limb was frequently evaluated for progress of resolution and healed well over the next 6 months without requiring any further immunosuppression or surgery.
| Discussion | ▴Top |
PG is a rare ulcerating lesion of unknown etiology. It usually begins as an inflamed papule or nodule which frequently ulcerates. This lesion is characterized by a necrotic or gangrenous base, and a raised violaceous undermined edge. PG does not have any unique or specific histological features; however, it is characterized by epidermal and dermal necrosis and neutrophilic infiltrates which can help support the diagnosis. It has also been shown to demonstrate pathergy, whereby lesions develop at sites of trauma.
There is no pathognomonic histological features for PG. In patients initially diagnosed with PG, there has been a reported misdiagnosis rate of up to 10% [2].
Epidemiological features of PG include a worldwide distribution affecting mostly the age group of 20 - 50 with women being more often affected than men [3]. The general incidence has been estimated to be between 3 and 10 per million per year [4].
PG is an uncommon cutaneous manifestation of SLE. More than 50% of patients with PG have an associated systemic disease, most commonly inflammatory bowel disease (14-34%) [3, 5]. However the proportion of patients with inflammatory bowel disease who develop PG is small. In a cohort study of 2,402 patients with inflammatory bowel disease, PG was detected in only 0.75% of patients [6].
The reason for the development of the inflammatory process that leads to PG is unclear. Neutrophils are prominent in biopsies from PG, and one study found abnormalities in neutrophil trafficking in patients with PG [7].
The sterile nature of the disease, with lack of infectious process, its often association with other autoinflammatory disorders, and its effective clinical response with steroids and immunosuppression does point towards an immunological etiology.
This case demonstrates the development of PG in a male patient who previously had no cutaneous features of SLE. The patient had been on regular low dose steroids and still developed PG. He was treated with increased steroid dose (prednisolone 60 mg) and meticulous wound hygiene and dressings, and the ulcers resolved with atrophic scarring within 6 months. The patient did not require any further immunosuppression or surgical intervention. Wound dressings that promote a moist wound environment and do not adhere to the wound base are preferred, as they may be beneficial for healing [8]. In patients with severe disease the mainstay treatment is systemic glucocorticoid therapy. Additional immunosuppressants such as cyclosporine can be added for those that cannot tolerate high dose steroids. Other immunosuppressants have also been shown to have a role in extensive disease such as infliximab, which is supported by a randomized controlled trial of 30 adults with PG, many of whom were concomitantly treated with topical or systemic therapy (primarily oral prednisolone). At the start of the trial, patients were given either a single 5 mg/kg infusion of infliximab (n = 13) or placebo (n = 17). After 2 weeks, significantly more patients in the infliximab group had clinical signs of improvement (46% versus 6%) [9].
The role of surgery is controversial in PG due to the possibility of pathergy. Surgical procedure is avoided in most cases. However, surgery may be necessary in a case of PG that runs the risk of necrosis and spreading infection. In terms of prognosis, studies have shown that with treatment, over half the patients with PG achieve remission within 1 year. In a series of 86 patients with PG, 68% of patients achieved complete remission within 6 months, and 95% were in remission within 3 years [5].
Interestingly, this patient did have a diagnosis of secondary antiphospholipid syndrome from his SLE. From the few documented cases of PG with SLE, the majority had antiphospholipid syndrome, e.g. Schmid et al [10], and it was believed that the PG association with SLE was as a result from secondary antiphospholipid syndrome as there had been an increased association with cutaneous ulcers including PG.
However there have also been case reports of PG in antiphospholipid antibody (aPL) negative SLE patients [11].
Many features of this rare disease such as etiology, pathogenesis and risk factors remain poorly understood. The association between SLE and PG is rare. A recent study by a medical analysis website in May 21, 2014 showed that from 358 people who have PG, three patients (0.84%) have lupus. They amount to 0.01% of all the 41,770 people who have lupus [12]. Further study is necessary to validate a relationship between SLE and PG.
| References | ▴Top |
- Binus AM, Qureshi AA, Li VW, Winterfield LS. Pyoderma gangrenosum: a retrospective review of patient characteristics, comorbidities and therapy in 103 patients. Br J Dermatol. 2011;165(6):1244-1250.
doi pubmed - Weenig RH, Davis MD, Dahl PR, Su WP. Skin ulcers misdiagnosed as pyoderma gangrenosum. N Engl J Med. 2002;347(18):1412-1418.
doi pubmed - von den Driesch P. Pyoderma gangrenosum: a report of 44 cases with follow-up. Br J Dermatol. 1997;137(6):1000-1005.
doi pubmed - Powell FC, Schroeter AL, Su WP, Perry HO. Pyoderma gangrenosum: a review of 86 patients. Q J Med. 1985;55(217):173-186.
pubmed - Bennett ML, Jackson JM, Jorizzo JL, Fleischer AB
Jr , White WL, Callen JP. Pyoderma gangrenosum. A comparison of typical and atypical forms with an emphasis on time to remission. Case review of 86 patients from 2 institutions. Medicine (Baltimore). 2000;79(1):37-46.
doi - Farhi D, Cosnes J, Zizi N, Chosidow O, Seksik P, Beaugerie L, Aractingi S,
et al . Significance of erythema nodosum and pyoderma gangrenosum in inflammatory bowel diseases: a cohort study of 2402 patients. Medicine (Baltimore). 2008;87(5):281-293.
doi pubmed - Adachi Y, Kindzelskii AL, Cookingham G, Shaya S, Moore EC, Todd RF
3rd , Petty HR. Aberrant neutrophil trafficking and metabolic oscillations in severe pyoderma gangrenosum. J Invest Dermatol. 1998;111(2):259-268.
doi pubmed - Miller J, Yentzer BA, Clark A, Jorizzo JL, Feldman SR. Pyoderma gangrenosum: a review and update on new therapies. J Am Acad Dermatol. 2010;62(4):646-654.
doi pubmed - Brooklyn TN, Dunnill MG, Shetty A, Bowden JJ, Williams JD, Griffiths CE, Forbes A,
et al . Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55(4):505-509.
doi pubmed - Schmid MH, Hary C, Marstaller B, Konz B, Wendtner CM. Pyoderma gangrenosum associated with the secondary antiphospholipid syndrome. Eur J Dermatol. 1998;8(1):45-47.
pubmed - Waldman MA, Callen JP. Pyoderma gangrenosum Preceding the diagnosis of systemic lupus erythematosus. Dermatology. 2005;210(1):64-67.
doi pubmed - Ehealth Me Review: could Pyoderma gangrenosum cause Lupus? http://www.ehealthme.com/print/cs4143432 2014.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.