

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 6, Number 9, September 2015, pages 420-425
Anesthetic Care of a Child With Propionic Acidemia
Mahmood Rafiqa, Joseph D. Tobiasa, b, c, d
aDepartment of Anesthesiology & Pain Medicine, Nationwide Children’s Hospital, Columbus, OH, USA
bDepartment of Anesthesiology & Pain Medicine, The Ohio State University College of Medicine, Columbus, OH, USA
cDepartment of Pediatrics, Division of Critical Care, Nationwide Children’s Hospital and The Ohio State University College of Medicine, Columbus, OH, USA
dCorresponding Author: Joseph D. Tobias, Department of Anesthesiology & Pain Medicine, Nationwide Children’s Hospital, 700 Children’s Drive, Columbus, OH 43205, USA
Manuscript accepted for publication August 13, 2015
Short title: Anesthesia and Propionic Acidemia
doi: http://dx.doi.org/10.14740/jmc2263w
| Abstract | ▴Top |
Propionic acidemia (PA), an autosomal recessive inborn error of metabolism, is one of the most frequent organic acidurias. The primary defect is deficiency of the mitochondrial enzyme, propionyl-CoA carboxylase which plays a key role in the final catabolism of the branched chain amino acids, the end-products of the β-oxidation of fatty acids, and the metabolism of the cholesterol side chains. Derangements in these pathways result in the accumulation of toxic metabolites which result in progressive end-organ dysfunction. Clinical manifestations include episodes of severe metabolic ketoacidosis, intercurrent infection, vomiting, hypotonia, convulsions, developmental delay with central nervous system involvement, gastroesophageal reflux, osteoporosis, pancreatitis, and cardiomyopathy. We present a 14-year-old girl with PA who required anesthetic care for magnetic resonance imaging (MRI) of the thoracolumbar spine. Previous reports of anesthetic care for these patients are reviewed, the end-organ involvement is discussed, and options for anesthetic care are presented.
Keywords: Propionic acidemia; Organic acidemia; Mitochondrial disorder
| Introduction | ▴Top |
Propionic acidemia (PA), an autosomal recessive inborn error of metabolism with an estimated incidence of 1 in 350,000, is one of the most frequent organic acidurias [1-3]. The condition is caused by deficiency of the mitochondrial enzyme, propionyl-CoA carboxylase (PCC) which plays a key role in the final catabolism of the branched chain amino acids (leucine, isoleucine, valine, threonine, and methionine), the end-products of the β-oxidation of odd chain fatty acids, and the metabolism of the cholesterol side chains [1-3]. Derangements in these pathways result in the accumulation of potentially toxic metabolites including propionic acid and methylmalonic acid [3]. Clinical manifestations include episodes of severe metabolic ketoacidosis, intercurrent infection, vomiting, hypotonia, convulsions, developmental delay with central nervous system (CNS) involvement, gastroesophageal reflux, osteoporosis, pancreatitis, and cardiomyopathy [1-4]. Definitive diagnosis requires assaying PCC activity in leukocytes and fibroblasts. Because of the significant genetic heterogeneity, the clinical picture varies widely from severe early-onset forms manifesting during the first days of life to mild late-onset variations, which may not be definitely diagnosed until adulthood. Treatment involves avoiding prolonged fasting, enteral feeding with a restriction of specific amino acids, the administration of biotin which acts as a cofactor for enzyme PCC, and supplementation with L-carnitine.
Patients with PA may require anesthesia for central line insertion, radiologic imaging, gastrostomy tube placement, scoliosis correction, and hepatic transplantation. We present a 14-year-old girl with PA who required anesthetic care for magnetic resonance imaging (MRI) of the thoracolumbar spine. Previous reports of anesthetic care for these patients are reviewed, the end-organ involvement is discussed, and options for anesthetic care are presented.
| Case Report | ▴Top |
Institutional Review Board approval is not required for case reports involving a single patient at Nationwide Children’s Hospital (Columbus, OH). A 14-year-old, 43.1 kg, wheelchair bound girl with PA required anesthetic care during an MRI of the thoracolumbar spine. Associated co-morbid conditions included traumatic brain injury, spasticity, scoliosis, encephalomalacia, osteopenia, and constipation. She was born at term via normal delivery. Her developmental milestones were normal up to the age of 3 years. She was diagnosed with PA after being hospitalized at 3 years of age for a brain injury following a fall from the stairs. At that time, she remained obtunded for 1 week and an MRI of the brain revealed bilateral globus pallidus injury with encephalomalacia. The family history was significant for six other family members with PA; however, their clinical manifestations were milder as none of them had any neurologic sequelae. After diagnosis, her medical management included a low protein diet to reduce amino acid catabolism and the administration of biotin, vitamin D3, and L-carnitine. The L-carnitine was subsequently discontinued due to gastrointestinal (GI) involvement including diarrhea resulting in dehydration. She had been hospitalized on many occasions due to recurrent clavicular fracture due to osteopenia, constipation, and poor oral intake. Preoperative vital signs revealed a blood pressure of 119/72 mm Hg, a heart rate of 100 beats/min, and a respiratory rate of 24 breaths/min. Physical examination revealed a thin girl with joint contractures, sitting in a wheelchair. Airway examination was not feasible given her cognitive state. Her cardiac and respiratory examinations were unremarkable. Preoperative laboratory results including complete blood count, electrolytes, renal function (BUN and creatinine), and hepatic function (ALT, AST, and bilirubin) were within normal range. A recent cardiac echocardiogram showed normal cardiac structure and function. The patient was held nil per os for 2 h for clear liquids and for 6 h for G-tube feedings. The patient was transported to the induction room of the MRI suite where standard American Society of Anesthesiologists’ monitors were placed. Anesthesia was induced by the inhalational of incremental concentrations of sevoflurane in 50% nitrous oxide and oxygen. After a sufficient depth of anesthesia was achieved, a peripheral intravenous cannula was placed and maintenance fluids started with 5% dextrose in 0.45% normal saline (70 mL/h). A blood glucose value was 120 mg/dL using a point-of-care testing device. A bolus dose of propofol (1.5 mg/kg) was administered and a size 3 laryngeal mask airway was placed without difficulty. Anesthesia was maintained with 2-3% sevoflurane in 50% oxygen and air while the patient breathed spontaneously. Care was taken to ensure ample padding of all pressure points as the patient was transferred from the induction cart to the MRI scanner. Anti-emetic prophylaxis included dexamethasone (4 mg) and ondansetron (4 mg). Morphine (2 mg) was administered to prevent emergence agitation. The imaging procedure required 1 h 20 min. The LMA was removed in the post-anesthesia care unit (PACU) when the patient was awake. Blood glucose was 145 mg/dL in the PACU. The patient was transferred to the inpatient ward after staying in the PACU for approximately 1 h. She was discharged home from the hospital later that same day approximately 3 - 4 h after the imaging procedure once she was able to tolerate her usual diet.
| Discussion | ▴Top |
PA is one of the most common organic acidemias resulting from deficiency or defective function of the enzyme, PCC which with the vitamin cofactor, biotin, converts propionyl-CoA to D-methylmalonyl CoA. Propionyl-CoA is normally formed in the catabolism of isoleucine, valine, methionine, threonine, odd-chain fatty acids, cholesterol side chains, thymine, and uracil. In normal individuals, methylmalonyl-CoA is subsequently converted to succinyl-CoA and shunted to the citric acid cycle. However, defective function of the enzyme, PCC, leads to an accumulation of propionic acid and other organic acids including 3-hydroxypropionate, methyl-citrate, propionyl-glycine, and tiglylglycine which result in end-organ toxicity.
The clinical presentation of PA is categorized into two subgroups: early-onset and late-onset PA. The vast majorities of patients with early-onset disease are born at full term and have unremarkable Apgar scores [5]. One-quarter of patients may have a family history of the unexplained death of a sibling or a sibling known to be affected by PA [5]. As with any autosomal recessive disorder, there may be a history of consanguinity. The early-onset disease is often more severe and presents with hypotonia, lethargy, vomiting, feeding difficulties, and tachypnea, during the first weeks of life, but as late as 3 months of life [5-7]. Other less common signs and symptoms include seizures, hepatomegaly, diarrhea, obstipation, coma, apnea, and hypothermia. Acute decompensation is marked by episodes of ketoacidosis and/or hyperammonemia, usually beginning in infancy. The late-onset form of PA is of milder severity with a median age of presentation of 16 months in one series [5, 7]. However, patients may present as early as 3 - 6 months after birth, but occasionally not until they are well into adulthood. The late-onset form is also more often associated with atypical presentations with a wide range of signs and symptoms. These signs and symptoms are often subtle and/or non-specific, including failure to thrive, feeding difficulties, acute or chronic encephalopathy, osteoporosis, neutropenia, thrombocytopenia, and immunodeficiency syndromes. The definitive diagnosis of PA requires an assay of PCC activity in leukocytes, liver, and most commonly skin fibroblast culture, the gold standard for diagnosis.
The medical management of PA is based on the pathophysiological process involved in the disease with avoidance of fasting and a low protein diet. The latter should be devoid of the specific amino acids (valine, isoleucine, threonine, and methionine) that lead to the production of propionic acid. Although such therapy with protein restriction may decrease end-organ effects of the disease, the extreme protein restriction may lead to failure to thrive and growth retardation [7]. Dietary protein restrictions need not be as strict for patients with late-onset disease. Dietary supplementation with L-carnitine and biotin may also be helpful.
Given the progressive, multi-organ system involvement of the disorder, anesthetic care may be required during a variety of surgical or imaging procedures. Table 1 summarizes previous reports from the literature regarding the perioperative care of these patients including two which describe the perioperative management during hepatic transplantation to treat the enzymatic defect responsible for the disorder [8-12].
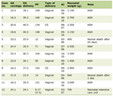 Click to view | Table 1. Previous Reports of Anesthetic Care for Patients With Propionic Acidemia |
As with any anesthetic care, the primary step is a thorough preoperative evaluation to identify end-organ involvement from PA as well as other co-morbid and acute conditions which may impact the perioperative course. End-organ involvement that impacts perioperative care may affect the cardiac, respiratory, and CNS. Cardiomyopathy and sudden cardiac death have been described as complications of several metabolic disorders including PA [13-16]. Cardiac involvement with cardiogenic shock may be the presenting symptom of PA [17]. Cardiac involvement is postulated to be the result of the direct toxic effects of propionic acid and its metabolites or secondary deficiencies of carnitine or selenium [2, 15, 18]. Acute cardiac decompensation can also occur in association with episodic metabolic decompensation and acidosis. In addition to effects on contractility, there are anecdotal reports of prolongation of the QT interval and sudden death [19, 20]. Preoperative assessment should generally include echocardiography and 12-lead electrocardiogram. Cardiac involvement may manifest as diffuse dilatation of the heart chambers, a hypokinetic left ventricle, and thickening of the ventricular walls. Cardiac involvement with depressed function may dictate alterations in the choice of agents for induction and maintenance of anesthesia as well as decisions regarding intraoperative monitoring of cardiac function with arterial and central venous access or transesophageal echocardiography especially for prolonged surgical procedures. We have previously reviewed the perioperative care of patients with prolonged QT syndrome [21]. A useful reference for the anesthesia provider regarding the effects of medications on the QT interval can be found at www.qtdrugs.com.
Another common co-morbid involvement that may result in perioperative complications is hypotonia affecting both the upper airway and the muscles of respiration. Such involvement may be difficult to assess during the preoperative assessment especially in patients with cognitive impairment and an inability to fully cooperative with preoperative pulmonary function testing. Preoperative assessment with identification of patients with an abnormal respiratory pattern, a history of recurrent pneumonia, the presence of gastroesophageal reflux, or swallowing problems may identify patients with an increased risk for perioperative respiratory complications. Preoperative preparation should include the aggressive treatment of respiratory infections and as cognitive function permits, instruction regarding the use of techniques such as incentive spirometry with the consideration of extubation to non-invasive ventilation techniques [22, 23]. The latter may be particularly useful following major or prolonged surgical procedures. Given the potential impact of long-acting anesthetic and neuromuscular blocking agents on postoperative respiratory function, we would recommend the use of short-acting agents. One of the case reports outlines the use of remifentanil while several used either desflurane or sevoflurane intraoperatively. When neuromuscular blockade is required, short-acting agent should be used with assurance that reversal is complete prior to tracheal extubation. Regardless of the anesthetic agents chosen, close monitoring of postoperative respiratory function is suggested.
A wide variety of neuropsychiatric and cognitive abnormalities have been described in patients with early-onset PA, being a major cause of morbidity associated with the disease. Specific considerations for the perioperative care of patients with seizures have been revised elsewhere [21]. Neuropathological findings include spongiform cerebral and cerebellar white-matter changes, along with degeneration in the putamen, thalamus, and globus pallidus [24-26]. Bilateral globus pallidus encephalomalacia was noted on the MRI in our patient. Neurological injury may be manifested as slow reactivity, pedaling movements, myoclonic jerks, large-amplitude tremors, axial hypotonic, and limb hypertonia. Axial hypotonia is responsible for a weak cough reflex and difficulty in clearing of airway secretions, which increases the risk of developing perioperative atelectasis and pneumonia (see above). The cognitive impairment associated with early-onset PA is generally development delay of varying severity, but acute or chronic encephalopathy with coma may also be seen. In early onset-disease, CNS involvement may manifest as respiratory problems (dyspnea or apnea), feeding problems (anorexia, difficulties with feeding, or repeated emesis), or depressed level of arousal (somnolence or lethargy). The neuropsychiatric and cognitive abnormalities in patients with the late-onset form of the diseases appear less frequently and are less severe as was the case with our patient. She was home schooled and was currently at the eighth grade level.
Understanding the patient’s baseline status and avoiding events that may precipitate metabolic crisis and acidosis remain priorities during the perioperative care of these patients. Triggers of metabolic decompensation may include fasting, dehydration, hypoglycemia, hypoxemia, and hypotension. Relatively minor infections may lead to metabolic decompensation and hence, elective surgery should not proceed if there is evidence of coexisting infection. On the day of surgery, prolonged fasting should be avoided as it may result in hypoglycemia, acidosis and metabolic decompensation. As was done in our patient, we limited the NPO time to 2 h with the administration of glucose containing clear liquids up to 2 h prior to the imaging procedure. Once intravenous access is obtained, 5-10% dextrose in isotonic fluids should be provided at maintenance to 1.5 times maintenance rates to provide 4 - 6 mg/kg/min of glucose and prevent hypoglycemia and catabolism. Periodic serum glucose monitoring is suggested.
There remains a paucity of information in the literature to determine the optimal anesthetic regimen for patients with PA. Theoretically, medications that are metabolized to propionic acid, odd chain organic acids, odd-chain alcohols, acrylic acid, or odd-chain fatty acids may initiate metabolic compromise as they are further metabolized to propionic acid, which can inhibit the urea and citric acid cycles. As such, it may be prudent to avoid neuromuscular blocking agents that are metabolized by ester hydrolysis including succinylcholine, cis-atracurium, atracurium, and mivacurium. Metabolites of these medications include odd-chain organic molecules [27-29]. Furthermore, succinylcholine may lead to hyperkalemia in myopathic conditions [30]. Although it has been used without adverse effects in a limited number of cases, there may be theoretical concerns with propofol as it is administered as an aqueous solution containing soybean oil, which is high in polyunsaturated fats, which may also be metabolized to propionic acid. While its use is acceptable for the induction of anesthesia, prolonged infusions of propofol should likely be avoided. Several medications in the non-steroidal anti-inflammatory class including naproxen, fenoprofen, ketoprofen, flurbiprofen, and oxaprozin are derived from propionic acid.
Intraoperatively, it has been suggested that lactated ringers (LR) should be avoided as lactate-containing fluids may not only contribute to the patient’s acid load, but also may be poorly metabolized by dysfunctional mitochondria. However, a more recent of 11 patients with various organic acidemias undergoing 19 anesthetics reported no adverse impact related to the intraoperative administration of LR [31]. Given the potential protein load, resuscitation with albumin containing fluids is also theoretically contraindicated. Another potential source of protein is the accumulation of blood in the stomach. For procedures involving oral or GI blood loss, a throat pack and nasogastric tube should be placed. For prolonged procedures, invasive arterial monitoring may be indicated to provide access to periodically check acid-base status, serum lactate, electrolytes, blood glucose, and ammonia.
In summary, PA is one of the most frequent of the organic acidurias resulting from a defect in the enzyme, PCC. The perioperative management of such patients focuses on the preoperative identification of end-organ involvement including decreased myocardial function, hypotonia involving the respiratory system and upper airway, as well as primary involvement of the CNS. Catastrophic metabolic decompensation may occur in the perioperative period. Postoperatively, patients should be observed for signs of clinical deterioration or respiratory insufficiency. For prolonged or complex surgical procedures, monitoring of acid-base status and glucose may be indicated to identify early decompensation.
| References | ▴Top |
- Wolf B, Hsia YE, Sweetman L, Gravel R, Harris DJ, Nyhan WL. Propionic acidemia: a clinical update. J Pediatr. 1981;99(6):835-846.
doi - Sass JO, Hofmann M, Skladal D, Mayatepek E, Schwahn B, Sperl W. Propionic acidemia revisited: a workshop report. Clin Pediatr (Phila). 2004;43(9):837-843.
doi - Deodato F, Boenzi S, Santorelli FM, Dionisi-Vici C. Methylmalonic and propionic aciduria. Am J Med Genet C Semin Med Genet. 2006;142C(2):104-112.
doi pubmed - Burlina AB, Dionisi-Vici C, Piovan S, Saponara I, Bartuli A, Sabetta G, Zacchello F. Acute pancreatitis in propionic acidaemia. J Inherit Metab Dis. 1995;18(2):169-172.
doi pubmed - Lehnert W, Sperl W, Suormala T, Baumgartner ER. Propionic acidaemia: clinical, biochemical and therapeutic aspects. Experience in 30 patients. Eur J Pediatr. 1994;153(7 Suppl 1):S68-80.
doi pubmed - Saudubray JM, Touati G, Delonlay P, Jouvet P, Schlenzig J, Narcy C, Laurent J, et al. Liver transplantation in propionic acidaemia. Eur J Pediatr. 1999;158(Suppl 2):S65-69.
doi pubmed - van der Meer SB, Poggi F, Spada M, Bonnefont JP, Ogier H, Hubert P, Depondt E, et al. Clinical outcome and long-term management of 17 patients with propionic acidaemia. Eur J Pediatr. 1996;155(3):205-210.
doi pubmed - Harker HE, Emhardt JD, Hainline BE. Propionic acidemia in a four-month-old male: a case study and anesthetic implications. Anesth Analg. 2000;91(2):309-311.
pubmed - Kim TW, Hall SR. Liver transplantation for propionic acidaemia in a 14-month-old male. Paediatr Anaesth. 2003;13(6):554-556.
doi - Karagoz AH, Uzumcugil F, Celebi N, Canbay O, Ozgen S. Anaesthetic management of a 2-year-old male with propionic adicemia. Pediatric Anesth. 2006;16:1290-1291.
doi pubmed - Ryu J, Shin YH, Ko JS, Gwak MS, Kim GS. Intractable metabolic acidosis in a child with propionic acidemia undergoing liver transplantation -a case report. Korean J Anesthesiol. 2013;65(3):257-261.
doi pubmed - Arcas-Bellas JJ, Arevalo-Ludena J, Onate ML, Aranzubia M, Alvarez-Rementeria R, Munoz-Alameda L. General anesthesia in an adult female with propionic acidemia: anesthetic considerations. Minerva Anestesiol. 2013;79(3):313-315.
pubmed - Kohlschutter A, Hausdorf G. Primary (genetic) cardiomyopathies in infancy. A survey of possible disorders and guidelines for diagnosis. Eur J Pediatr. 1986;145(6):454-459.
doi pubmed - Guertl B, Noehammer C, Hoefler G. Metabolic cardiomyopathies. Int J Exp Pathol. 2000;81(6):349-372.
doi pubmed - Massoud AF, Leonard JV. Cardiomyopathy in propionic acidaemia. Eur J Pediatr. 1993;152(5):441-445.
doi - Ou P, Touati G, Fraisse A, Sidi D, Kachaner J, Saudubray JM, Bonnet D. [A rare cause of cardiomyopathy in childhood: propionic acidosis. Three case reports]. Arch Mal Coeur Vaiss. 2001;94(5):531-533.
pubmed - Laemmle A, Balmer C, Doell C, Sass JO, Haberle J, Baumgartner MR. Propionic acidemia in a previously healthy adolescent with acute onset of dilated cardiomyopathy. Eur J Pediatr. 2014;173(7):971-974.
doi pubmed - Mardach R, Verity MA, Cederbaum SD. Clinical, pathological, and biochemical studies in a patient with propionic acidemia and fatal cardiomyopathy. Mol Genet Metab. 2005;85(4):286-290.
doi pubmed - Kakavand B, Schroeder VA, Di Sessa TG. Coincidence of long QT syndrome and propionic acidemia. Pediatr Cardiol. 2006;27(1):160-161.
doi pubmed - Jameson E, Walter J. Cardiac arrest secondary to long QT(C )in a child with propionic acidemia. Pediatr Cardiol. 2008;29(5):969-970.
doi pubmed - Kako H, Martin DP, Cartabuke R, Beebe A, Klamar J, Tobias JD. Perioperative management of a patient with Rett syndrome. Int J Clin Exp Med. 2013;6(5):393-403.
pubmed - Jaber S, Michelet P, Chanques G. Role of non-invasive ventilation (NIV) in the perioperative period. Best Pract Res Clin Anaesthesiol. 2010;24(2):253-265.
doi pubmed - Pelosi P, Jaber S. Noninvasive respiratory support in the perioperative period. Curr Opin Anaesthesiol. 2010;23(2):233-238.
doi pubmed - Haas RH, Marsden DL, Capistrano-Estrada S, Hamilton R, Grafe MR, Wong W, Nyhan WL. Acute basal ganglia infarction in propionic acidemia. J Child Neurol. 1995;10(1):18-22.
doi pubmed - Surtees RA, Matthews EE, Leonard JV. Neurologic outcome of propionic acidemia. Pediatr Neurol. 1992;8(5):333-337.
doi - Feliz B, Witt DR, Harris BT. Propionic acidemia: a neuropathology case report and review of prior cases. Arch Pathol Lab Med. 2003;127(8):e325-328.
pubmed - Kisor DF, Schmith VD. Clinical pharmacokinetics of cisatracurium besilate. Clin Pharmacokinet. 1999;36(1):27-40.
doi pubmed - Stiller RL, Cook DR, Chakravorti S. In vitro degradation of atracurium in human plasma. Br J Anaesth. 1985;57(11):1085-1088.
doi pubmed - Savarese JJ, Ali HH, Basta SJ, Embree PB, Scott RP, Sunder N, Weakly JN, et al. The clinical neuromuscular pharmacology of mivacurium chloride (BW B1090U). A short-acting nondepolarizing ester neuromuscular blocking drug. Anesthesiology. 1988;68(5):723-732.
doi pubmed - Martyn JA, Richtsfeld M. Succinylcholine-induced hyperkalemia in acquired pathologic states: etiologic factors and molecular mechanisms. Anesthesiology. 2006;104(1):158-169.
doi - Ruzkova K, Weingarten TN, Larson KJ, Friedhoff RJ, Gavrilov DK, Sprung J. Anesthesia and organic aciduria: is the use of lactated Ringer's solution absolutely contraindicated? Paediatr Anaesth. 2015;25(8):807-817.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.