

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 7, Number 12, December 2016, pages 531-535
Cardiac Amyloidosis Presenting With Myocardial Ischemia: Case Report and Literature Review
Francisco Alvarez Kellera, d, Peiman Jamshidia, Bart De Boecka, Katharina Glatzb, Miriam Nowackc
aCardiology Department, Heart Center, Lucerne Hospital, Switzerland
bPathology Department, University Hospital Basel, Switzerland
cPathology Department, Lucerne Hospital, Switzerland
dCorresponding Author: Francisco Alvarez Keller, Cardiology Department, Heart Center, Lucerne Hospital, Switzerland
Manuscript accepted for publication September 16, 2016
Short title: Cardiac Amyloidosis Presenting With Myocardial Ischemia
doi: https://doi.org/10.14740/jmc2648w
| Abstract | ▴Top |
We present a 65-year-old woman with a history of exercise-induced retrosternal pain and dyspnea. Both coronary angiography and CMR imaging showed evidence of microvascular dysfunction. After several admissions with acute coronary syndrome and heart failure, a myocardial biopsy was performed which revealed abundant AL amyloid deposits. We present this case in detail and review articles.
Keywords: Cardiac amyloidosis; Myocardial ischemia; Microvascular dysfunction; Heart failure
| Introduction | ▴Top |
Cardiac amyloidosis is caused by deposition of an insoluble protein material, known as amyloid substance, in the cardiac interstitium. AL amyloidosis is the most commonly diagnosed form of cardiac amyloidosis. It is associated with a B-cell dyscrasia and AL amyloid fibrils are composed of fragments of either kappa or lambda monoclonal immunoglobulin light chains. In some patients, overt multiple myeloma may co-exist and is associated with a poorer prognosis [1]. One or many vital organ systems may be involved (commonly the kidneys, liver, peripheral and autonomic nervous systems and soft tissues). The heart is frequently the predominant organ affected by amyloid deposition and is the sole organ involved in a small proportion of patients [2].
Although AL amyloidosis typically is viewed as a rare disease, its incidence is similar to that of Hodgkin’s lymphoma or chronic myelogenous leukemia [3]. It is estimated to affect 5 - 12 people per million per year, although autopsy studies suggest that the incidence might be higher [4].
The condition usually presents as a restrictive cardiomyopathy that leads to death from heart failure in most patients. Diagnosis at early stages of the disease and prompt initiation of therapy are crucial.
| Case Report | ▴Top |
A 65-year-old Caucasian female was referred to our cardiology clinic with a history of exercise-induced retrosternal pain and dyspnea. She denied any history of smoking or alcohol/drug abuse and was not on any medications. On physical examination, vital signs were normal. The remainder of the examination was unremarkable, in particular no signs of heart failure. Laboratory values on admission revealed elevated inflammatory and cardiac markers. A 12-lead electrocardiography showed a bifascicular block and T-wave inversion in the anterior chest wall leads (Fig. 1). A transthoracic echocardiogram revealed moderate concentric left ventricular hypertrophy with preserved global left ventricular systolic function, infero-lateral apical hypokinesis, impaired relaxation and elevated filling pressures. A coronary angiography was performed which revealed no epicardial coronary artery stenosis (Fig. 2).
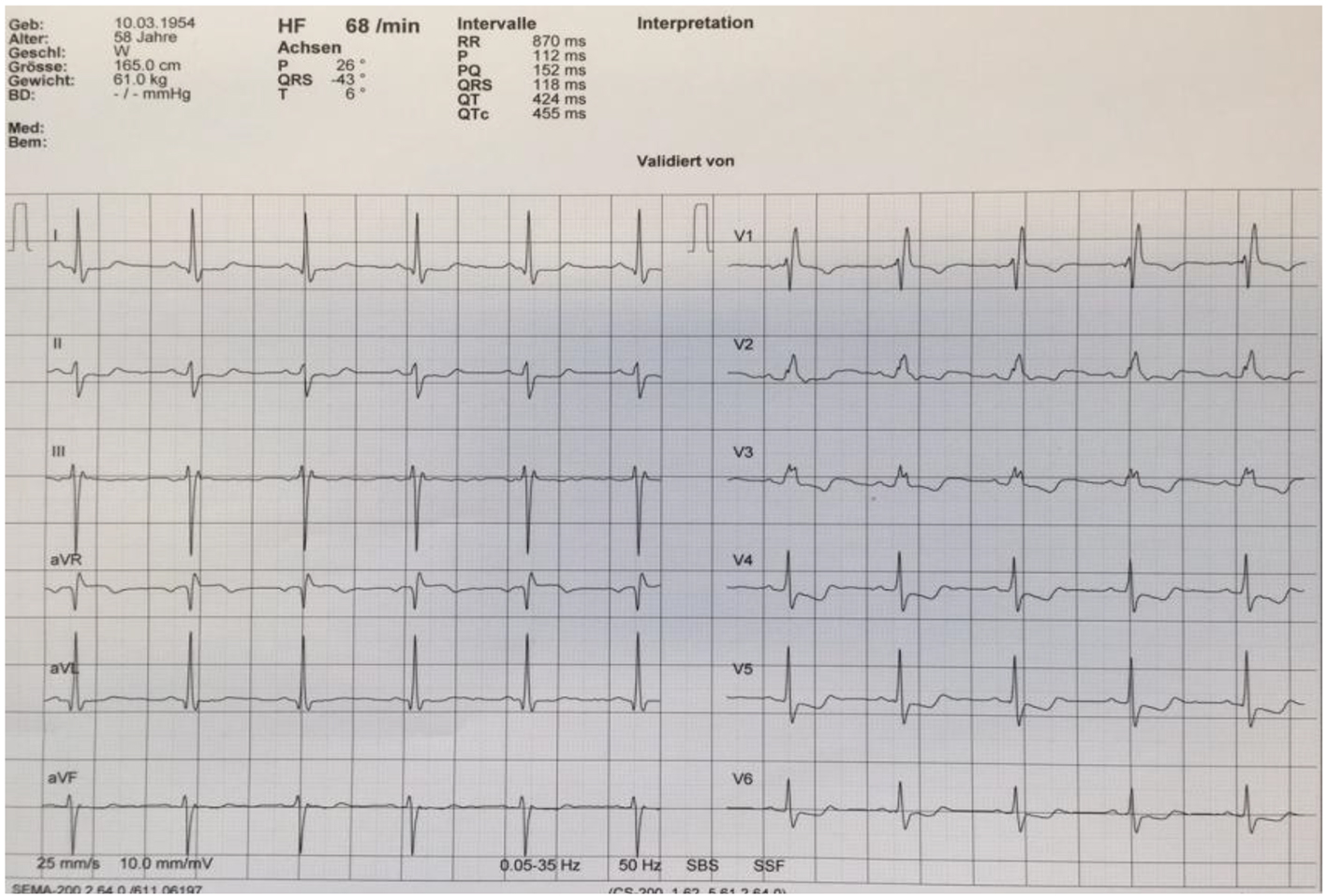 Click for large image | Figure 1. Electrocardiogram reveals a right bundle branch block and left anterior fascicular block, in addition T-wave inversion in leads V3-V5. |
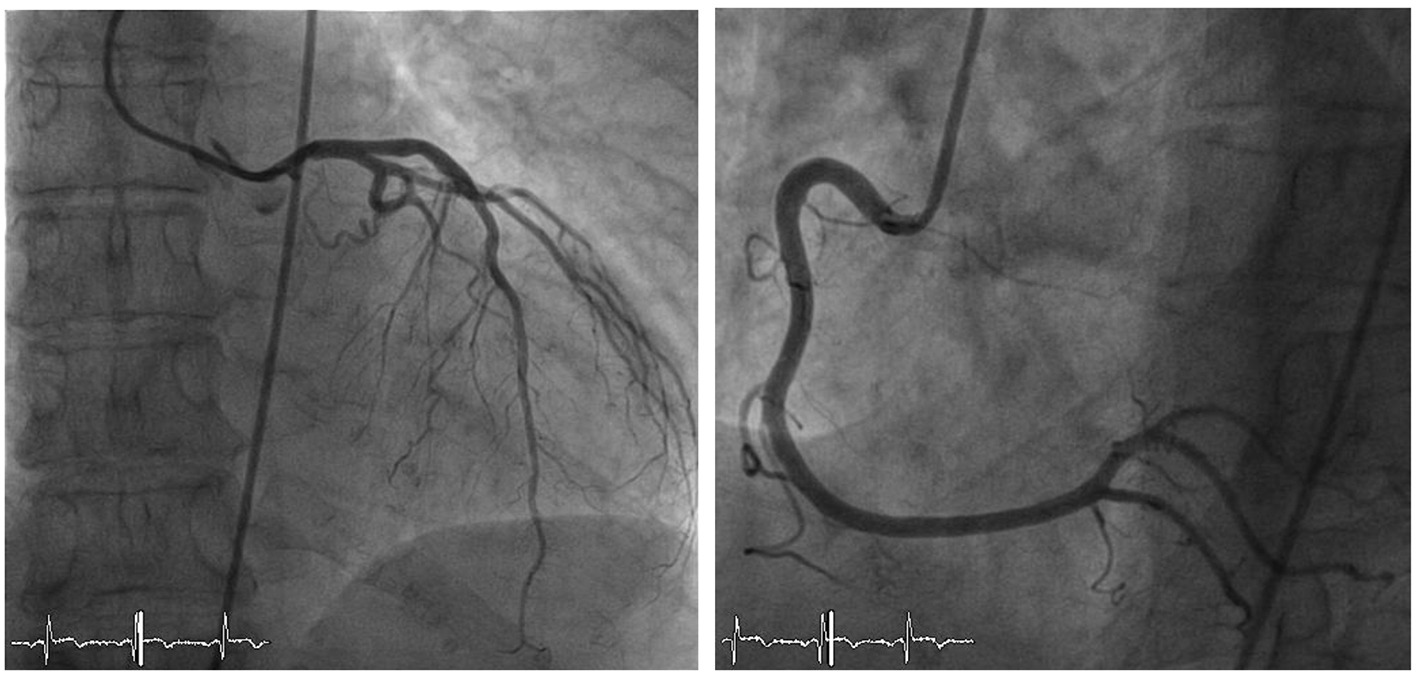 Click for large image | Figure 2. Coronary angiography showing normal left and right coronary arteries. |
The clinical picture was initially interpreted as perimyocarditis. Therapy was therefore started with diclofenac. Cardiac amyloidosis was also considered; however, the protein electrophoresis was unremarkable, in particular no monoclonal gammopathy.
The follow-up took place 1 month later. The patient was still complaining of exercise-induced pectanginous symptoms. A two-dimensional transthoracic echocardiogram revealed a normal global left ventricular function (EF 58%) with persistence of inferolateral hypokinesis. For further investigation, a cardiac MRI adenosine stress test was carried out. This revealed no indications of myocarditis or myocardial fibrosis. A relevant infarction was excluded. The left ventricular myocardium showed borderline thickening and a detectable endocardial delay in perfusion in almost all sections of the wall (Fig. 3). Microvascular dysfunction or cardiac syndrome X was initially postulated and a therapy was started with angiotensin-converting enzyme (ACE) inhibitors and beta-blocker.
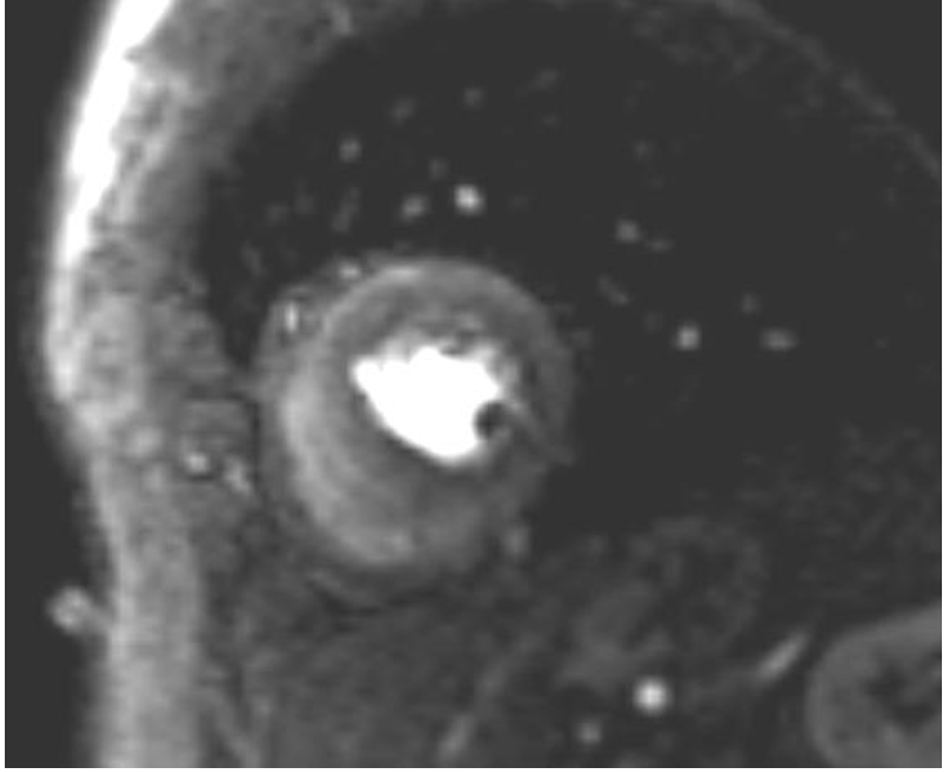 Click for large image | Figure 3. MRI adenosine stress test showing diffuse endocardial perfusion delay. |
Over the further course of treatment, the patient suffered relapses resulting in several hospitalizations due to cardiac decompensation. Over time, she developed a severe heart failure. A follow-up revealed an LVEF of 35% with diffuse hypokinesis. Impaired relaxation and elevated filling pressures with restrictive mitral inflow pattern were consistent with severe diastolic dysfunction. Also noted was moderate to severe mitral regurgitation. An additional coronary angiography was carried out. It still revealed normal epicardial coronary arteries with clear slow flow at the level of the intramyocardial vessels (myocardial blush) as a sign of possible microvascular dysfunction. No improvement of the symptoms was achieved in spite of intensification of the anti-ischemic therapy with nicorandil, nebivolol and isosorbide dinitrate.
Right heart catheterization with endomyocardial biopsy was performed during her last hospitalization, which took place about 15 months after the onset of symptoms. In addition to elevated pulmonary arterial pressures, it showed the dip-and-plateau sign noted on right ventricular pressure tracings. The biopsy revealed abundant AL amyloid deposits (Fig. 4). The bone marrow biopsy revealed plasma cell infiltration of 70%, leading to the diagnosis of amyloidosis associated with AL kappa multiple myeloma. In addition, the amyloidosis was found to have affected the liver (hepatomegaly), kidneys (proteinuria), joints and soft tissue (macroglossia and suspected carpal tunnel syndrome). There was still no visible M component in the protein electrophoresis, but hypogammaglobulinemia was detected.
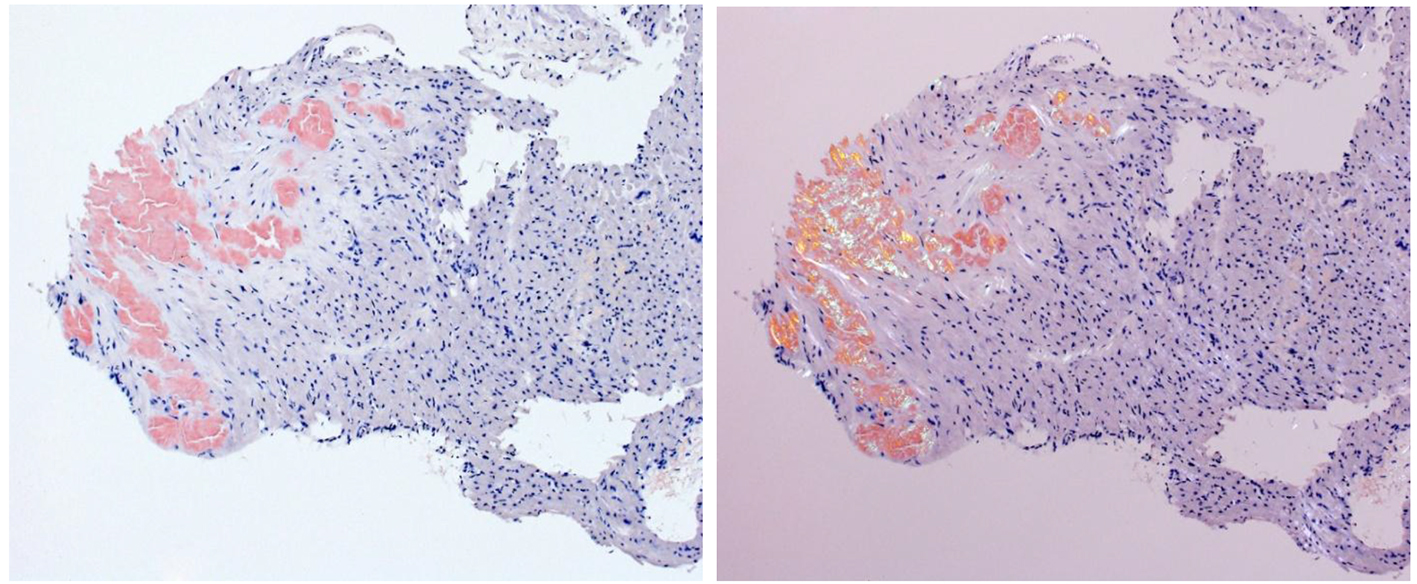 Click for large image | Figure 4. Endomyocardial biopsy: the amyloid deposits turn red in the Congo red staining (left); same cut of Congo red staining in polarized light; abnormal green to yellow birefringence of amyloid deposits (right). |
Chemotherapy was started with bortezomib and dexamethasone which had to be discontinued due to adverse reactions. The patient opted for a supportive and palliative care and expired on the 45th day of hospitalization.
The autopsy of the patient revealed kappa-type plasmocytoma with moderate bone marrow infiltration and generalized systemic AL amyloidosis as cause of death (Fig. 5).
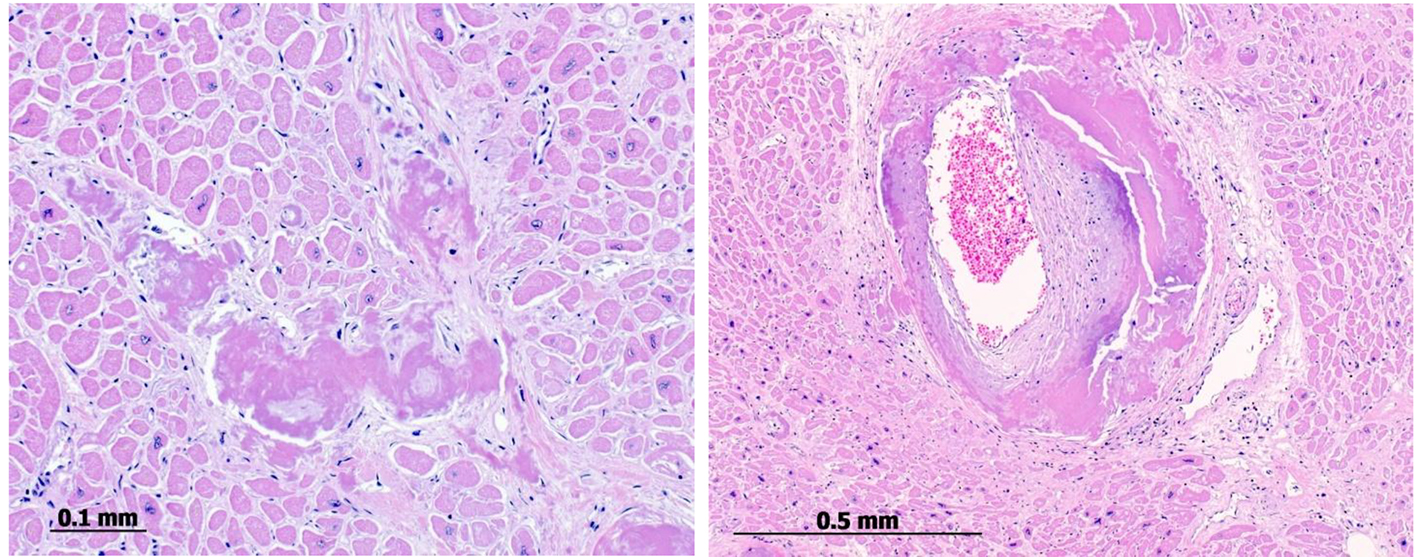 Click for large image | Figure 5. Postmortem endomyocardial histological samples show abundant interstitial, subendocardial (left) and small artery media (right) deposits of amyloid substance. |
| Discussion | ▴Top |
The typical patient with heart failure resulting from AL amyloidosis frequently presents with rapidly progressive signs and symptoms. Progressive dyspnea is common, almost always associated with evidence of elevated right-sided filling pressure. Peripheral edema may be profound, and in late-stage disease, ascites is not uncommon. Weight loss, which is common, may represent the effects of the systemic disease or may be a manifestation of cardiac cachexia. Patients with cardiac amyloidosis may present with chest discomfort. Usually this is not typical of angina and is associated with congestive heart failure. However, typical angina can occur because of involvement of the small vessels of the heart. Characteristic electrocardiogram features include low voltage in the limb leads or loss of anterior forces consistent with anteroseptal infarction. There have been case reports of intramural coronary artery stenosis and obstruction in patients with amyloidosis and concurrent heart failure, angina, or arrhythmias, but many patients with infarct patterns on electrocardiogram do not have major underlying atherosclerotic disease. These infarct patterns may be caused by amyloid-mediated occlusion of smaller intramyocardial arteries. Atrial fibrillation and heart block are also common electrocardiogram findings. When present, symptomatic conduction system disease often follows the onset of congestive heart failure and may require pacemaker placement. Imaging studies may be positive, leading to cardiac catheterization with apparently normal epicardial coronary arteries on coronary angiography. Myocardial flow reserve in such patients is impaired because of the small vessel involvement, and a small but persistent elevation in serum troponin may be present, leading to a misdiagnosis of non-Q-wave infarction. Presumably, the troponin elevation represents ongoing myocyte necrosis and it has been shown to be a negative prognostic factor. Small vessel cardiac amyloid may occur in the absence of wall thickening on the echocardiogram, although there is often a mild elevation of left ventricular filling pressure suggesting diastolic abnormalities of the ventricle. This presentation of amyloidosis is rare; it is seen in only 1-2% of patients with cardiac involvement [5].
The diagnosis of AL amyloidosis requires demonstration of amyloid in tissue and demonstration of a plasma cell dyscrasia. Tissue amyloid deposits demonstrate apple-green birefringence when stained with Congo red and viewed under polarizing microscopy. Once a tissue diagnosis of amyloidosis has been established, confirmation of AL disease requires demonstration of a plasma cell dyscrasia by a bone marrow biopsy showing predominance of λ- or κ-producing plasma cells or by the presence of a monoclonal light chain in the serum or urine [6]. The recently introduced serum free-light-chain (FLC) assay, a nephelometric immunoassay, has a sensitivity for circulating FLCs that is reportedly > 10-fold that of immunofixation electrophoresis [7].
The treatment and prognosis of cardiac amyloidosis directly depend on the underlying etiology of amyloid deposition. However, in general calcium channel blockers are contraindicated due to case reports of worsening left ventricular function in patients with cardiac amyloidosis, thought to be related to their negative inotropic effects [8]. Beta blockers are often avoided for the same reason [9]. Digoxin should be used with caution because it is bound extracellularly by amyloid fibrils, which may cause patients with amyloidosis to be “hypersensitive” to its effects [10]. ACE inhibitors or other vasodilating agents are of varying utility depending on the presence or absence of comorbidities such as the nephrotic syndrome or refractory orthostatic hypotension. Pacemakers may be required depending on the degree of conduction system disease [8]. Some clinicians also advocate treating patients with warfarin with a goal international normalized ratio of 2.0 - 3.0 because of the higher incidence of intrachamber thrombi seen in cardiac amyloidosis, even in the absence of atrial fibrillation [11].
Several case reports and retrospective cohort studies have described a marginal benefit with the use of internal cardioverter defibrillators in patients with cardiac amyloidosis. Detection of early cardiac involvement and proper identification of patients at high risk for sudden cardiac death due to rapid progressive amyloidosis needs to be defined [12].
The prognosis for patients with AL amyloidosis is poor. Median survival is 13 months without treatment and can be extended to 17 months with cyclic oral melphalan and prednisone therapy [13, 14]. However, such treatment rarely results in complete remission of disease or reversal of end organ dysfunction because of amyloid deposits. Only 5% of patients survive for more than 10 years [15]. Recently, high-dose intravenous melphalan has been combined with autologous peripheral blood stem cell transplantation resulting in varying degrees of hematologic response, decreased end-organ dysfunction and prolonged survival in some patients [4, 16, 17]. High-dose thalidomide and dexamethasone have been tried as well, but few patients are able to tolerate the toxicity of this regimen [18]. Cardiac transplantation has been used in some patients with AL amyloidosis, but long-term survival is less favorable than in patients transplanted for other conditions [19-22].
This is an unusual case of cardiac amyloidosis with atypical clinical presentation and negative initial workup for amyloidosis, including serum/urine protein electrophoresis tests. Serum FLC assay was not performed as the index of suspicion for systemic amyloidosis was initially low. Both coronary angiography and CMR imaging showed evidence of microvascular dysfunction. This pattern may be caused by amyloid-mediated occlusion of smaller intramyocardial arteries.
In conclusion, patients with cardiac amyloidosis have very poor survival without therapy. Diagnosis at early stages of the disease and prompt initiation of therapy are crucial. Non-invasive diagnostic methods, such as echocardiography and cardiovascular MRI, are useful for diagnosis in patients with signs and symptoms of progressive heart failure, but some findings are not specific for cardiac amyloid and can be found in other restrictive cardiomyopathies.
A high index of suspicion for systemic amyloidosis warrants obtaining also a serum FLC assay as it can detect circulating FLC with 10-fold greater sensitivity than immunofixation in AL amyloidosis [7].
Endomyocardial biopsy is the only method by which the presence of cardiac amyloidosis can be directly verified but this method is invasive, expensive and not always available [23].
A rapid initial therapy mainly with diuretics is usually needed to stabilize patients with congestive heart failure. Conventional heart failure drugs are usually not well tolerated and the long-term benefits are uncertain. Chemotherapeutic agents could delay the progress of the disease and prolong survival in some patients, but rarely result in complete remission or reversal of end organ dysfunction. Cardiac transplantation, although still controversial, should be considered in highly selected cases.
Acknowledgments
I would like to give special thanks to Prof. K. Glatz, Pathology Department University Hospital Basel and Dr. M. Nowack, Pathology Department Lucerne Hospital for kindly supplying the histological images. I also want to express my special appreciation and thanks to Dr. P. Jamshidi and Dr. B. De Boeck for their support and guidance.
| References | ▴Top |
- Kyle RA, Greipp PR, O'Fallon WM. Primary systemic amyloidosis: multivariate analysis for prognostic factors in 168 cases. Blood. 1986;68(1):220-224.
pubmed - Dubrey SW, Cha K, Anderson J, Chamarthi B, Reisinger J, Skinner M, Falk RH. The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. QJM. 1998;91(2):141-157.
doi pubmed - Gertz MA, Lacy MQ, Dispenzieri A. Amyloidosis. Hematol Oncol Clin North Am. 1999;13(6):1211-1233, ix.
doi - Skinner M, Sanchorawala V, Seldin DC, Dember LM, Falk RH, Berk JL, Anderson JJ, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med. 2004;140(2):85-93.
doi pubmed - Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation. 2005;112(13):2047-2060.
doi pubmed - Sanchorawala V. Light-chain (AL) amyloidosis: diagnosis and treatment. Clin J Am Soc Nephrol. 2006;1(6):1331-1341.
doi pubmed - Kumar S, Dispenzieri A, Katzmann JA, Larson DR, Colby CL, Lacy MQ, Hayman SR, et al. Serum immunoglobulin free light-chain measurement in primary amyloidosis: prognostic value and correlations with clinical features. Blood. 2010;116(24):5126-5129.
doi pubmed - Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med. 1997;337(13):898-909.
doi pubmed - Gertz MA, Falk RH, Skinner M, Cohen AS, Kyle RA. Worsening of congestive heart failure in amyloid heart disease treated by calcium channel-blocking agents. Am J Cardiol. 1985;55(13 Pt 1):1645.
doi - Rubinow A, Skinner M, Cohen AS. Digoxin sensitivity in amyloid cardiomyopathy. Circulation. 1981;63(6):1285-1288.
doi pubmed - Pellikka PA, Holmes DR, Jr., Edwards WD, Nishimura RA, Tajik AJ, Kyle RA. Endomyocardial biopsy in 30 patients with primary amyloidosis and suspected cardiac involvement. Arch Intern Med. 1988;148(3):662-666.
doi pubmed - Halwani O, Delgado DH. Cardiac amyloidosis: an approach to diagnosis and management. Expert Rev Cardiovasc Ther. 2010;8(7):1007-1013.
doi pubmed - Skinner M, Anderson J, Simms R, Falk R, Wang M, Libbey C, Jones LA, et al. Treatment of 100 patients with primary amyloidosis: a randomized trial of melphalan, prednisone, and colchicine versus colchicine only. Am J Med. 1996;100(3):290-298.
doi - Kyle RA, Gertz MA, Greipp PR, Witzig TE, Lust JA, Lacy MQ, Therneau TM. A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N Engl J Med. 1997;336(17):1202-1207.
doi pubmed - Kyle RA, Gertz MA, Greipp PR, Witzig TE, Lust JA, Lacy MQ, Therneau TM. Long-term survival (10 years or more) in 30 patients with primary amyloidosis. Blood. 1999;93(3):1062-1066.
pubmed - Sanchorawala V, Wright DG, Seldin DC, Dember LM, Finn K, Falk RH, Berk J, et al. An overview of the use of high-dose melphalan with autologous stem cell transplantation for the treatment of AL amyloidosis. Bone Marrow Transplant. 2001;28(7):637-642.
doi pubmed - Gertz MA, Lacy MQ, Dispenzieri A, Gastineau DA, Chen MG, Ansell SM, Inwards DJ, et al. Stem cell transplantation for the management of primary systemic amyloidosis. Am J Med. 2002;113(7):549-555.
doi - Palladini G, Perfetti V, Perlini S, Obici L, Lavatelli F, Caccialanza R, Invernizzi R, et al. The combination of thalidomide and intermediate-dose dexamethasone is an effective but toxic treatment for patients with primary amyloidosis (AL). Blood. 2005;105(7):2949-2951.
doi pubmed - Hosenpud JD, Uretsky BF, Griffith BP, O'Connell JB, Olivari MT, Valantine HA. Successful intermediate-term outcome for patients with cardiac amyloidosis undergoing heart transplantation: results of a multicenter survey. J Heart Transplant. 1990;9(4):346-350.
pubmed - Pelosi F, Jr., Capehart J, Roberts WC. Effectiveness of cardiac transplantation for primary (AL) cardiac amyloidosis. Am J Cardiol. 1997;79(4):532-535.
doi - Hosenpud JD, DeMarco T, Frazier OH, Griffith BP, Uretsky BF, Menkis AH, O'Connell JB, et al. Progression of systemic disease and reduced long-term survival in patients with cardiac amyloidosis undergoing heart transplantation. Follow-up results of a multicenter survey. Circulation. 1991;84(5 Suppl):III338-343.
pubmed - Dubrey SW, Burke MM, Khaghani A, Hawkins PN, Yacoub MH, Banner NR. Long term results of heart transplantation in patients with amyloid heart disease. Heart. 2001;85(2):202-207.
doi pubmed - McCarthy RE, 3rd, Kasper EK. A review of the amyloidoses that infiltrate the heart. Clin Cardiol. 1998;21(8):547-552.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.