

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 8, Number 6, June 2017, pages 180-182
Joubert Syndrome: A Rare Case Report From Saudi Arabia
Khulood M. ALHarbi
Department of Pediatrics, College of Medicine, Taibah University, Medina, Saudi Arabia
Manuscript accepted for publication May 10, 2017
Short title: Joubert Syndrome
doi: https://doi.org/10.14740/jmc2832w
| Abstract | ▴Top |
Joubert syndrome (JS) is a rare genetic disorder. We report a 15-month-old girl who presented with developmental delay, decrease vision, failure to thrive and history of two hospital admissions with respiratory distress and recurrent aspiration pneumonia due to gastroesophageal reflux disease. On examination, she had facial dysmorphism, nystagmus, retinopathy and hypotonia. Brain magnetic resonance imaging revealed cerebellar vermis hypoplasia, abnormally thick superior cerebellar peduncles, a “batwing” configuration of the fourth ventricle, and the characteristic molar tooth sign of mid-brain suggestive of JS. Patient was referred to the genetic department outside our hospital where the diagnosis of JS was confirmed. In conclusion, awareness of the characteristic clinical and radiological findings in JS will help in early diagnosis, appropriate counseling and proper rehabilitation.
Keywords: Joubert syndrome; Genetic disorder; Respiratory distress; Molar tooth sign
| Introduction | ▴Top |
Joubert syndrome (JS) is a rare autosomal recessive genetic heterogeneously inherited disorder, named after Marie Joubert in 1969 [1]. It is characterized by congenital ataxia, hypotonia, developmental delay and at least one of the following features: neonatal respiratory disturbances and abnormal eye movements, including nystagmus and oculomotor apraxia. It can be identified on axial magnetic resonance imaging (MRI) as the molar tooth sign (MTS), which is considered as an essential finding for the diagnosis of JS [2, 3].
The prevalence of JS is less than 1 in 100,000, and by 2009 only 200 cases have been reported all over the world [4]. Because of its non-specific clinical presentation, its correct diagnosis is usually not made for a long period up to years after birth, even though the clinical features are present very early in life [5]. Maria et al reported 33 months as an average age for its diagnosis [6]. Most children with this syndrome survive infancy to reach adulthood [7].
Early detection of this condition is essential so that appropriate intervention can be started as early as possible.
| Case Report | ▴Top |
A 43-day-old girl, outcome of term uneventful pregnancy delivered by cesarean section, with no complications, birth weight of 2.5 kg, NICU admission for a day due to neonatal jaundice, and no history of abnormal breathing pattern, presented to the emergency department in our hospital with history of cough and shortness of breath with recurrent choking and cyanosis during feeding. She was admitted to the hospital with respiratory distress diagnosed as aspiration pneumonia and gastroesophageal reflux disease (massive GERD) confirmed later on by barium swallow study. Patient received antibiotic for 7 days and improved and discharged on omeprazole.
At age of 3 months, the patient was admitted again to the hospital with similar problem of respiratory distress diagnosed as pneumonia and treated with antibiotic and discharged in good condition to be followed with the gastroenterologist for GERD management.
Patient missed frequent appointment and was seen again at age of 15 months in the out patient clinic with main complaint of developmental delay, decrease vision and failure to thrive.
There is consanguinity between the parents; the patient has one older healthy sister, and no history of similar cases in the family. Parents noticed that their daughter was unable to keep up with developmental milestones compared to her older sister and she was not gaining weight.
On examination, the patient was alert, but inconsistently focusing visually. Intermittent eye movements to extremes of gaze were noted. She showed facial dysmorphic features in the form of forehead prominence, deep-set eyes, hypertelorism, depressed nasal bridge, posteriorly rotated low set ears, triangular mouth and low frontal hairline, and overlapping toes, no polydactyly or syndactyly. Her weight and height were below 3rd percentile and head circumference was normal for age.
There were multiple cafe au lait spots on her abdomen, no organomegaly. Cardiac and respiratory examination were normal on auscultation. Neurological examination revealed hypotonia with hyporeflexia.
Laboratory investigations, including CBC, blood sugar, electrolytes, C-reactive protein, liver enzymes, urea and creatinine, TSH, free T4 and T3 were all normal.
Brain MRI done showed cerebellar vermis hypoplasia, thick elongated superior cerebellar peduncles, a “batwing” configuration of the fourth ventricle, and molar tooth appearance of mid brain suggestive of JS (Fig. 1).
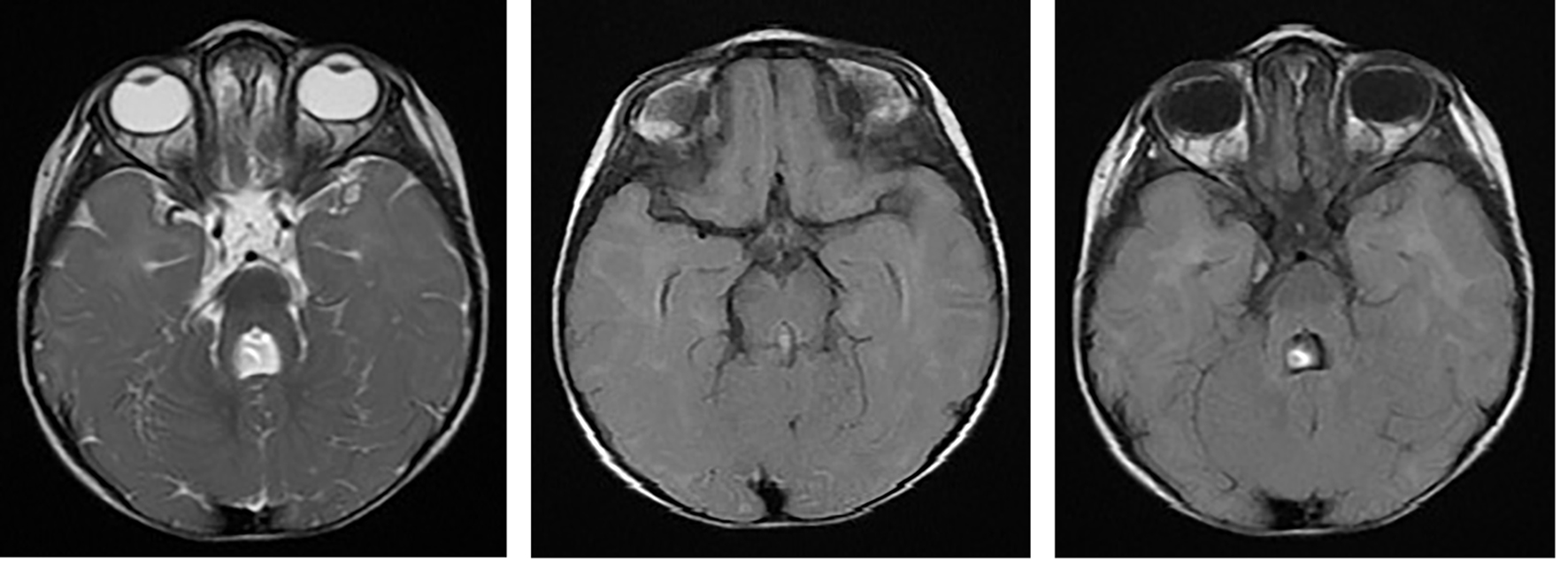 Click for large image | Figure 1. Axial brain MRI image showing cerebellar vermis hypoplasia, thick elongated superior cerebellar peduncles, a “batwing” configuration of the fourth ventricle, a molar tooth sign of mid brain. |
Echocardiography revealed no abnormality. Abdomen ultrasound was negative for a kidney or liver involvement. Ophthalmology examination showed decreased visual acuity, retinopathy but normal optic disc. Hearing assessment was normal. Genetic analysis was not available in our hospital. Patient was referred to the genetic department outside our hospital where the diagnosis of JS was confirmed.
Extensive genetic counseling for the parents was done. Patient was now followed with multidisciplinary team (behavior and development, physiotherapy, pediatric neurology, nephrology, ophthalmology) and frequent ultrasound for the abdomen. She has been involved in early intervention program including speech, occupational and physical therapy.
| Discussion | ▴Top |
The clinical features most frequently reported as important for the diagnosis of JS are hypotonia during infancy, mental retardation/developmental delay (in particular language and motor skills), in addition to one or both of the following that helped in diagnosis: abnormal breathing pattern (hyperpnea and/or apnea, sometimes alternating) and abnormal eye movements (nystagmus) [8]. The present case had all the clinical symptoms with the exception of mental retardation which may have been overlooked.
From radiologic point of view, the features necessary for a diagnosis of JS are the MTS on axial views from cranial MRI studies composed of three main findings: cerebellar vermis hypoplasia, deepened interpeduncular fossa, and thick, horizontal enlarged superior cerebellar peduncles [4]. This sign was apparent in the present case. Cerebellar vermis hypoplasia had been reported as a component of other disorders like trisomy 21, occipital encephalocele and dandy walker malformation [9, 10]. Also, hypotonia should be associated with other features to be considered as an essential clinical feature [10].
JS is classified into two main types, pure JS, and JS and related disorder (JSRD). The term JSRD refers to a group of disorders presenting the pathognomonic features of JS in association with multiple systemic congenital abnormalities. The prevalence of multicystic renal disease and lower survival rate are more observed among patients with retinal dystrophy compared to those without retinal dystrophy [11]. Ophthalmology examination of the present case showed retinopathy but normal optic disc with no evidence of renal involvement. Our patient had severe GERD but no association was found on literature review.
Prenatal diagnosis of JS has been documented by others [10, 12], and the ultrasonographic findings showed increased nuchal translucency.
The prognosis of JS cases is not bad, if discovered and managed early through implementing an intervention program for patients including special education, occupational, physical, and speech therapy. This program has shown significant benefits in advancement of developmental milestones for several patients with JS [13].
Up to our knowledge, this JS case is the first to be reported from Saudi Arabia, (AlMadinah region) and we present it to summarize main clinical and radiological mile stones for its diagnosis, draw attention of physicians to the presence of this syndrome and for the possibility of diagnosing it antenatally by ultrasonographic study.
| References | ▴Top |
- Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19(9):813-825.
doi pubmed - Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300). Eur J Hum Genet. 2007;15(5):511-521.
doi pubmed - Boltshauser E, Isler W. Joubert syndrome: episodic hyperpnea, abnormal eye movements, retardation and ataxia, associated with dysplasia of the cerebellar vermis. Neuropadiatrie. 1977;8(1):57-66.
doi pubmed - Choh SA, Choh NA, Bhat SA, Jehangir M. MRI findings in Joubert syndrome. Indian J Pediatr. 2009;76(2):231-235.
doi pubmed - Akcakus M, Gunes T, Kumandas S, Kurtoglu S, Coskun A. Joubert syndrome: Report of a neonatal case. Paediatr Child Health. 2003;8(8):499-502.
doi pubmed - Maria BL, Quisling RG, Rosainz LC, Yachnis AT, Gitten J, Dede D, Fennell E. Molar tooth sign in Joubert syndrome: clinical, radiologic, and pathologic significance. J Child Neurol. 1999;14(6):368-376.
doi pubmed - Bandichhode ST, Anitha MS, Pandav A. Joubert Syndrome-A Case Report. JKIMSU. 2013;2(2):138-140.
- Parisi MA, Glass IA. In: GeneReviews at GeneTests-GeneClinics: Medical Genetics Information Resource. Seattle: Copyright, University of Washington; 1997-2006. [last cited on 2010 Nov 1]. Joubert syndrome. Available from: http://www.geneclinics.org. or http://www.genetests.org. 2006.
- Satran D, Pierpont ME, Dobyns WB. Cerebello-oculo-renal syndromes including Arima, Senior-Loken and COACH syndromes: more than just variants of Joubert syndrome. Am J Med Genet. 1999;86(5):459-469.
doi - Aslan H, Ulker V, Gulcan EM, Numanoglu C, Gul A, Agar M, Ark HC. Prenatal diagnosis of Joubert syndrome: a case report. Prenat Diagn. 2002;22(1):13-16.
doi pubmed - Saraiva JM, Baraitser M. Joubert syndrome: a review. Am J Med Genet. 1992;43(4):726-731.
doi pubmed - al-Talabani JA, Shubbar AI, Sobkey E. Cranial sonographic findings in Joubert's syndrome. J Clin Ultrasound. 1999;27(1):41-44.
doi - Parisi MA. Clinical and molecular features of Joubert syndrome and related disorders. Am J Med Genet C Semin Med Genet. 2009;151C(4):326-340.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.