

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 8, Number 8, August 2017, pages 252-255
Atypical Presentation of Gordon Syndrome and Its Management: A Report of Three Patients
Sushil Mehandrua, Attiya Haroona, Vikas Singha, Avais Masuda, Eric J. Costanzoa, Mayurkumar Patela, Elmer Sadiang-Abaya, Arif Asifa, b
aDepartment of Medicine, Jersey Shore University Medical Center, Hackensack-Meridian Seton Hall School of Medicine, Neptune, NJ, USA
bCorresponding Author: Arif Asif, Department of Medicine, Seton Hall-Hackensack-Meridian School of Medicine, Jersey Shore University Medical Center, Neptune, NJ, USA
Manuscript submitted July 10, 2017, accepted July 24, 2017
Short title: Atypical Presentation of Gordon syndrome
doi: https://doi.org/10.14740/jmc2874w
| Abstract | ▴Top |
Gordon syndrome (GS) usually presents in children with hyperkalemia, hypertension and hyperchloremic metabolic acidosis. However, it has been reported in adults, where diagnosis can be missed in the absence of vigilance for electrolytes and acid base abnormalities. GS is traditionally managed with hydrochlorothiazide and restricted sodium diet. We report adult patients with GS, who were successfully treated with a potassium-restricted diet. Three female patients (aged 70, 61, and 79 years) presented to the clinic with untreated isolated hyperkalemia for several years. Patients did not complain of headache, palpitation, chest pain, polyuria or hematuria. Upon examination, two patients were normotensive and one patient had hypertension (150/90 mm Hg). Laboratory analysis revealed hyperkalemia (6.2, 6.1 and 6.2 mEq/L), serum bicarbonate level of 30, 27 and 28 mEq/L, hyperchloremia (108, 107 and 94 mEq/L) and serum normal creatinine (0.90, 0.8 and 0.78 mg/dL), based upon normal reference lab values. Plasma aldosterone and renin were normal. Based on patients’ history, physical examination, laboratory findings and absence of other causes of hyperkalemia such as the history of the use of NSAID, ACE inhibitors (angiotensin receptor blockers, aldosterone receptor blockers, and beta-blockers), and the absence of diabetes mellitus and other causes of hyporeninemic hypoaldosteronism, a diagnosis of GS was made. All three patients were treated with a restricted potassium diet contrary to a low sodium diet and hydrochlorothiazide. A 4-week follow-up showed normal serum potassium levels (4.2, 4.6 and 4.9 mEq/L) in all three patients. While GS has been reported previously in adults and can present with normal blood pressure, its management by a restricted potassium diet has not been reported, to the best of our knowledge. GS should be included in the differential diagnosis of adult patients presenting with unexplained hyperkalemia.
Keywords: Gordon syndrome; Pseudohypoaldosteronism; Hyperkalemia
| Introduction | ▴Top |
Pseudohypoaldosteronism (PHA) is an electrolyte abnormality, which is characterized by an apparent state of renal tubular unresponsiveness or resistance to the action of aldosterone [1]. PHA has been further sub-classified into PHA I or classical form and PHA II or Gordon syndrome (GS). PHA II or GS is a rare renal tubular defect characterized by hypertension, hyperkalemia and metabolic acidosis in the presence of normal glomerular filtration rate (GFR), low renin and normal aldosterone levels, reported both in children and adults [2, 3]. It is an autosomal dominant disease. The molecular basis for most individuals who have GS is linked to the loss-of-function mutations in WNK1 and WNK4 or KLHL3 and CUL3 mutations [4, 5]. Patients with GS are traditionally managed with dietary sodium restriction and hydrochlorothiazide (HCTZ) [6]. However, we report three patients who presented with GS as adults and their management with a low potassium diet contrary to a restricted sodium diet and HCTZ diuretic.
| Case Reports | ▴Top |
Case 1
A 70-year-old female was referred to the clinic for the workup of hyperkalemia (serum potassium 6.2 mEq/L). She was reported to have hyperkalemia for the past several years. Other laboratory data showed serum sodium level of 146 mEq/L and CO2 of 30 mEq/L. She did not complain of polyuria, oliguria, hematuria, weakness, palpitation, headache or dizziness. Her past medical history included aortic incompetence, mitral regurgitation and urethrocele. There was no family history of hypertension, diabetes, hyperkalemia or kidney failure. On examination, she was awake and alert, oriented × 3 with a blood pressure of 121/60 mm Hg and a heart rate of 70 beats per minute. There was no peripheral or periorbital edema, abdominal tenderness or swelling. Workup for hyperkalemia displayed low potassium excretion (urine potassium 18) in the presence of markedly elevated serum potassium levels (serum potassium 6. 2 mEq/L) (Table 1). A diagnosis of GS was made based on hyperkalemia, low potassium excretion, normal aldosterone, normal GFR (65 mL/min) and no history of NSAID overdose, ACEI abuse, diabetes mellitus and hypoaldosteronism. She was started on a low potassium diet as patient refused to take any medication including hydrochlorthiazide. Patient was already on a sodium-restricted diet. Four weeks follow-up showed a serum potassium level of 4.2 mEq/L (Table 2).
 Click to view | Table 1. Laboratory Parameters of Patients With GS Before Low Potassium Diet |
Click to view | Table 2. Laboratory Parameters of Patients With GS After Low Potassium Diet |
Case 2
A 61-year-old female was referred to the clinic for isolated hyperkalemia (serum potassium: 6.1 mEq/L) for the past several years. She had no other complaints. She had a past history of hyperlipidemia. There was no family history of hypertension, hyperkalemia or kidney disease. On examination, she had a blood pressure of 120/80 mm Hg and a heart rate of 75 beats per minute. Workup done for hyperkalemia showed urine potassium excretion of 7.5 mEq/L, serum potassium level of 5.8 mEq/L, GFR of 60 mL/min and normal aldosterone level (Table 1). Based on these laboratory findings, a diagnosis of GS was made and the patient was started on a restricted potassium diet. The patient refused to take any medication including hydrochlorthiazide. This patient was already on a sodium-restricted diet. Three weeks follow-up showed normal potassium level of 4.6 mEq/L without any medication (Table 2).
Case 3
A 79-year-old female presented with weakness and dizziness. She had no complaints of hematuria, polyuria, oliguria, edema leg or pain abdomen. She had a history of hypertension and insulin-dependent diabetes mellitus. There was no family history of hypertension, diabetes or kidney failure. On examination, she had a blood pressure of 150/90 mm Hg and a heart rate of 70 beats per minute. Workup done showed serum potassium level of 6.2 mEq/L, urine potassium excretion of 7 mEq/L, and a GFR of 70 mL/min (Table 1). Based on these laboratory findings, a diagnosis of GS was made and the patient was started on 25 mg HCTZ and a low potassium diet. The patient was already observing a sodium-restricted diet. Four weeks follow-up showed serum potassium level of 4.9 mEq/L, normal blood pressure (120/80 mm Hg) and no complaint of weakness (Table 2).
| Discussion | ▴Top |
According to a recent survey, 29.1% of USA population is suffering from hypertension [7]. Although 90-95% of these cases are due to essential hypertension, 5-10% cases are due to secondary causes [7]. Renal parenchyma and renal vascular disorder constitutes one of the most common causes of secondary hypertension [8]. GS is a rare cause of adult hypertension and hyperkalemia. Most patients of GS are asymptomatic until adolescence, when hypertension develops. However, GS can present with normal blood pressure and isolated hyperkalemia [9]. The three cases presented had hyperkalemia; however, only one had hypertension. All were following a sodium-restricted diet and all three responded to a potassium-restricted diet with resolution of hyperkalemia. Only one patient required HCTZ, which controlled hypertension in that patient.
Management of GS is based upon the understanding of the pathophysiology of this disorder. It has been postulated that hypertension in GS is caused by an increase in sodium reabsorption in the distal nephrons through gain of function of sodium chloride co-transporter (NCC) and activation of the OSR1/SPAK-NCC phosphorylation cascade [10]. There is an associated decrease in renal outer medullary potassium channels (ROMK) (decreased potassium excretion), increased epithelial sodium channels (ENaC) (enhanced sodium reabsorption) and increased para-cellular transport of chloride through tight junctions (Fig. 1). All these factors together contribute to hypertension, hyperkalemia and hyperchloremic metabolic acidosis. It is important to recognize that plasma renin level is usually low/normal. Plasma and urine aldosterone levels are usually normal because low renin tends to lower and hyperkalemia tends to increase aldosterone levels [10].
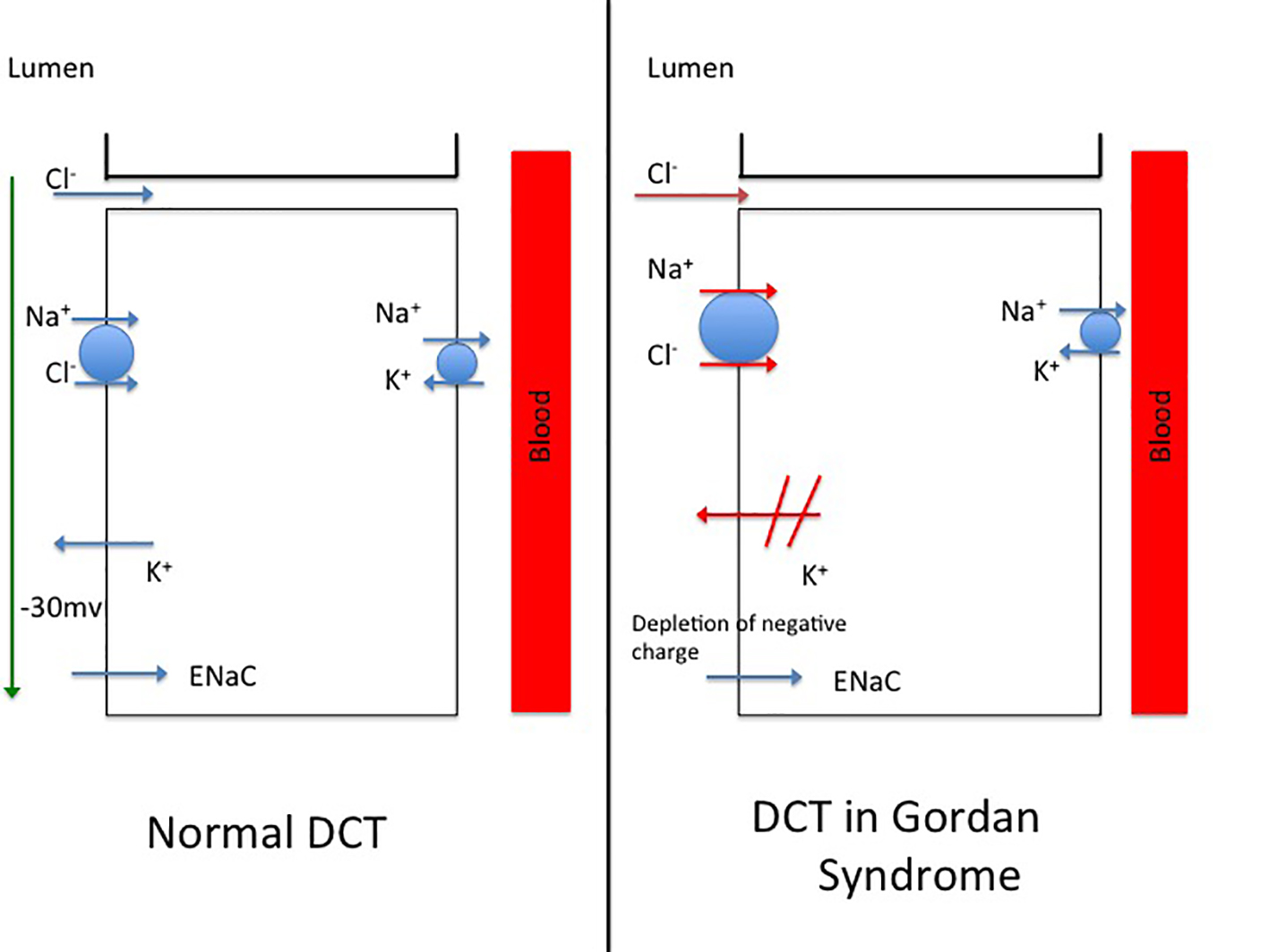 Click for large image | Figure 1. (a) A model of distal convoluted tubule (DCT) in a normal nephron. (B) Postulated mechanisms involved in the distal convoluted tubule of a patient with Gordon’s syndrome. |
Conventionally, patients with GS respond well to aggressive salt restriction and low dose thiazide diuretics [9]. Thiazide diuretics inhibit NCC and hence reduce sodium reabsorption. In a study by Mayan et al on eight affected patients with GS, marked sensitivity to HCTZ was noticed with normalization of blood pressure and sodium [11]. In contrast, two of our patients had normal blood pressure and refused to take HCTZ. All of our patients were already observing a low-sodium diet. Hence, we for the first time, prescribed a low-potassium diet to our three patients with GS. One of the patients had hypertension and agreed to take HCTZ. The sodium-restriction that this patient was observing did not help with hyperkalemia or hypertension. This patient responded well to a low-potassium diet and HCTZ. Follow-up laboratory results after 3 - 4 weeks in all three patients displayed correction of serum potassium level and normal blood pressure in the patient who also suffered from hypertension.
Conclusion
While GS has been reported previously in adults and can present with normal blood pressure, to the best of our knowledge, its management by restricted potassium diet has not been reported previously. GS should be included in the differential diagnosis of patients presenting with unexplained hyperkalemia.
Conflicts of Interest
None.
| References | ▴Top |
- Ellison DH. Pseudohypoaldosteronism Type II. 2011 Nov 10 [updated 2017 Feb 16].
- O’Shaughnessy KM. Gordon Syndrome: a continuing story. Pediatr Nephrol. 2015;30(11):1903-1908.
doi pubmed - Achard JM, Disse-Nicodeme S, Fiquet-Kempf B, Jeunemaitre X. Phenotypic and genetic heterogeneity of familial hyperkalaemic hypertension (Gordon syndrome). Clin Exp Pharmacol Physiol. 2001;28(12):1048-1052.
doi pubmed - Ohta A, Schumacher FR, Mehellou Y, Johnson C, Knebel A, Macartney TJ, Wood NT, et al. The CUL3-KLHL3 E3 ligase complex mutated in Gordon’s hypertension syndrome interacts with and ubiquitylates WNK isoforms: disease-causing mutations in KLHL3 and WNK4 disrupt interaction. Biochem J. 2013;451(1):111-122.
doi pubmed - Tsuji S, Yamashita M, Unishi G, Takewa R, Kimata T, Isobe K, Chiga M, et al. A young child with pseudohypoaldosteronism type II by a mutation of Cullin 3. BMC Nephrol. 2013;14:166.
doi pubmed - Gordon RD, Geddes RA, Pawsey CG, O’Halloran MW. Hypertension and severe hyperkalaemia associated with suppression of renin and aldosterone and completely reversed by dietary sodium restriction. Australas Ann Med. 1970;19(4):287-294.
pubmed - Tatiana Nwankwo, Sung Sug (Sarah) Yoon,Vicki Burt, Sc.M, Qiuping Gu. Hypertension among adults in the United States: national health and nutrition examination survey, 2011-2012. NCHS Data Brief No. 133. 2013.
- Kostakis ID, Tsoukalas NG, Aravantinos DC, Gkizis IG, Cholidou KG, Papadopoulos DP. A case report of Gordon’s syndrome in a 20-year-old male with free medical family history. Hellenic J Cardiol. 2013;54(1):64-68.
pubmed - Yang SS, Morimoto T, Rai T, Chiga M, Sohara E, Ohno M, Uchida K, et al. Molecular pathogenesis of pseudohypoaldosteronism type II: generation and analysis of a Wnk4(D561A/+) knockin mouse model. Cell Metab. 2007;5(5):331-344.
doi pubmed - Louis-Dit-Picard H, Barc J, Trujillano D, Miserey-Lenkei S, Bouatia-Naji N, Pylypenko O, Beaurain G, et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat Genet. 2012;44(4):456-460, S451-453.
- Mayan H, Vered I, Mouallem M, Tzadok-Witkon M, Pauzner R, Farfel Z. Pseudohypoaldosteronism type II: marked sensitivity to thiazides, hypercalciuria, normomagnesemia, and low bone mineral density. J Clin Endocrinol Metab. 2002;87(7):3248-3254.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.