

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 8, Number 9, September 2017, pages 283-286
A Rare Case of Surviving Aortic Dissection With Dominant Left Coronary in a Patient With Newly Diagnosed Marfan Syndrome
Nada Mohameda, c, Jiayi Mab, Nicole Lib, Allison Hallb
aDepartment of Internal Medicine, United Health Services, Johnson City, NY, USA
bSUNY Upstate Medical University, Syracuse, NY, USA
cCorresponding Author: Nada Mohamed, Department of Internal Medicine, United Health Services, Johnson City, NY, USA
Manuscript submitted August 11, 2017, accepted August 25, 2017
Short title: Aortic Dissection in Marfan Syndrome
doi: https://doi.org/10.14740/jmc2890w
| Abstract | ▴Top |
Marfan syndrome is an autosomal dominant disease with high penetrance and marked phenotypic variation caused by a mutation in the fibrillin-1 gene (FBN1) on chromosome 15. There is known multisystem involvement of Marfan syndrome; however, cardiovascular manifestations are the main cause of morbidity and mortality in affected patients. Although the most common cardiovascular manifestation is mitral valve prolapse, it is aortic involvement that has the greatest impact on prognosis. Within the aortic pathologies, aortic dilation is quite common, occurring in 60-80% of all adults with Marfan syndrome. Aortic dilation typically occurs in the aortic root due to its higher elastic fiber content, composed of fibrillin, and the wall stress and cyclic torsion that this segment experiences during ventricular systole. Aortic regurgitation, a late manifestation, is generally secondary to aortic dilation. In the general population, right coronary dominance is seen in 70%, whereas left dominance is found in approximately 18% with the remaining population demonstrating a balanced pattern. We present a patient with no prior medical history who presented with a 3-month history of cough and worsening shortness of breath, was admitted for pneumonia, was subsequently found to have an 83 mm (8.3 cm) dilation of the ascending aorta and aortic root, and survived in part due to left dominant coronary circulation.
Keywords: Marfan syndrome; Dominant left coronary; Aortic dissection; Aortic root dilatation
| Introduction | ▴Top |
First described in 1896 by French pediatrician Antoine Bernard-Jean Marfan in a 5-year-old girl, Marfan syndrome is an autosomal dominant disease with high penetrance and marked phenotypic variation caused by a mutation in the fibrillin-1 gene (FBN1) on chromosome 15. FBN1, the main component of microfibrils, has structural and regulatory functions in the extracellular matrix. Specifically, it controls the TGF-β signaling pathway. Mutated forms of FBN1 lead to excessive TGF-β signaling and activation resulting in the typical Marfan syndrome phenotype involving the ocular, skeletal, cardiovascular, pulmonary, cutaneous, and neurological systems.
Despite the multisystem involvement of Marfan syndrome, cardiovascular manifestations are the main cause of morbidity and mortality in affected patients. The most common cardiovascular manifestation is mitral valve prolapse, although aortic involvement has the greatest impact on prognosis as aortic dissection is the most feared complication of this disease. Current guidelines for management of valvular heart disease state that surgery should be considered in a patient with Marfan syndrome who has aortic root disease with a maximum ascending aortic diameter greater than or equal to 45 mm (4.5 cm).
We present a patient with no prior medical history who presented with a 3-month history of cough and worsening shortness of breath, was admitted for pneumonia, and was subsequently found to have an 83 mm (8.3 cm) dilation of the ascending aorta and aortic root.
| Case Report | ▴Top |
Our patient is a 31-year-old previous healthy Caucasian man standing at 6′5″ and weighing 190 lbs. He had no prior cardiac history and initially presented to the walk-in clinic with a 2-week history of shortness of breath. Pertinent positives included fatigue, productive cough and chest pain associated with the cough. Dyspnea was present at rest and worsened with exertion. Vital signs were stable except for tachycardia with a rate of 128 per minute. The patient was afebrile with a temperature of 97.6 °F and was oxygenating at 99% on room air. Labs done included complete blood count (CBC), comprehensive metabolic panel (CMP), thyroid function tests, and urine drug screen. All labs were within normal limits except the urine drug screen which was positive for cannabis and methamphetamines. A chest X-ray (CXR) performed at that time showed no evidence of pleural effusion or pneumothorax (Fig. 1). Approximately 6 weeks later, he returned to care as his symptoms had not improved, but in fact worsened. A CXR done showed bilateral pulmonary infiltrates suspicious for bilateral lower lobe pneumonia (Fig. 2). The initial plan was to admit the patient as an inpatient for IV antibiotics but the patient refused admission stating he would like to try outpatient oral antibiotics. He was discharged with a 7-day course of levofloxacin 750 mg qday with instructions to return if his symptoms worsened.
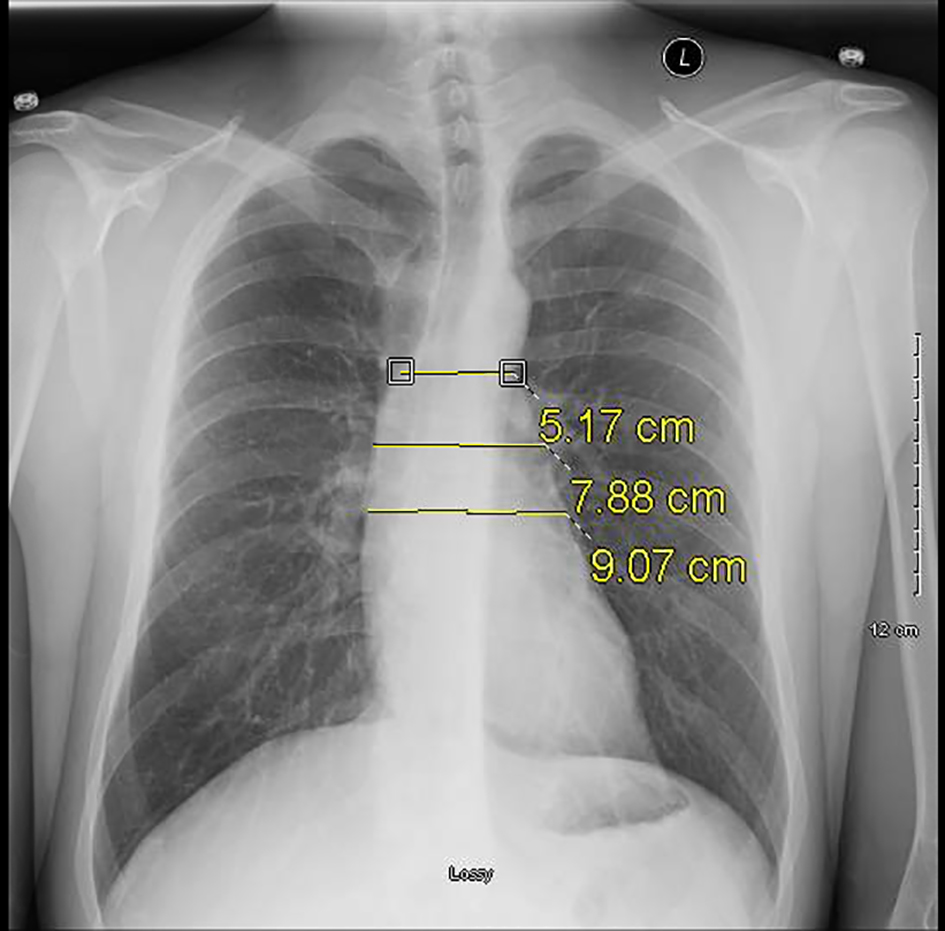 Click for large image | Figure 1. Serial plain radiograph images demonstrating progressively widening mediastinum on October 20, 2016. |
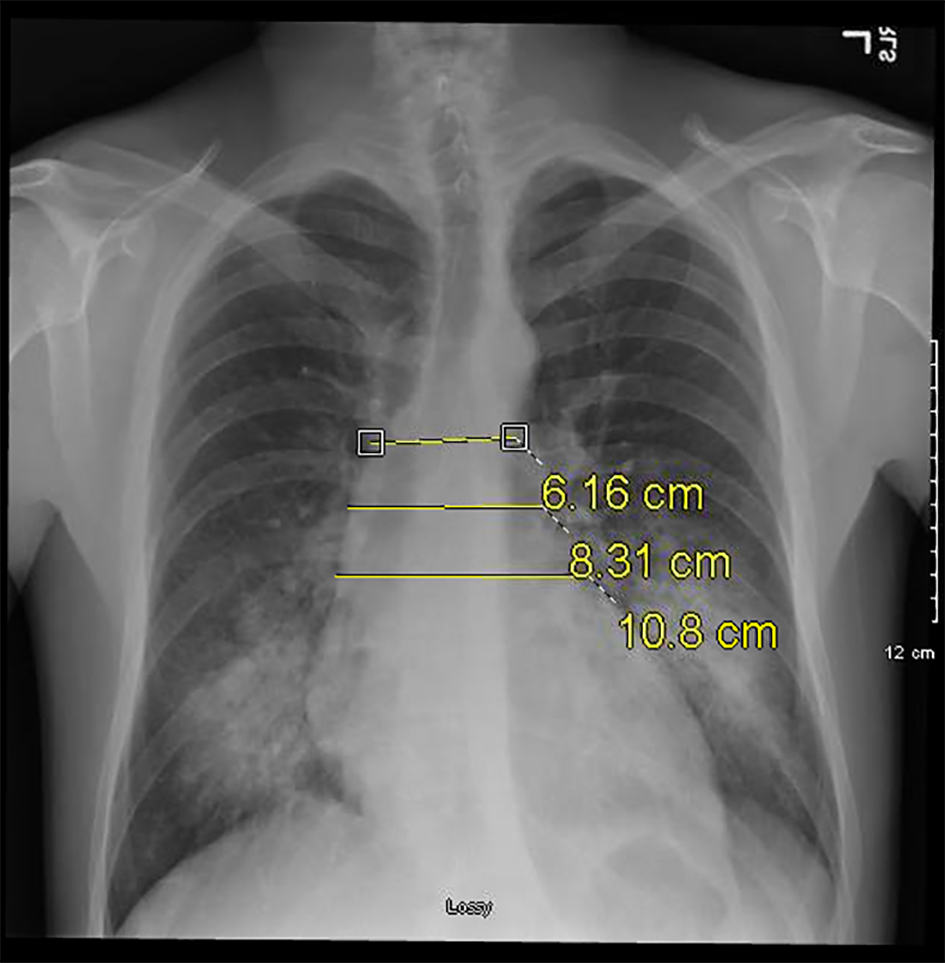 Click for large image | Figure 2. Serial plain radiograph images demonstrating progressively widening mediastinum on December 1, 2016. |
Two days following his second visit, the patient returned to the emergency room with worsening dyspnea. Upon admission, the patient was afebrile and saturating at 100% on room air. However, vital signs were significant for tachypnea with a respiratory rate of 26 and tachycardia with a heart rate of 122. Pulmonary exam was significant for diminished breath sounds bilaterally and dullness to percussion in both lung bases. A CXR done at that time revealed bilateral pulmonary infiltrates similar to the imaging done 2 days prior (Fig. 3). The patient was admitted and started on IV piperacillin-tazobactam, azithromycin, and vancomycin. Shortly thereafter the patient experienced cardiac arrest necessitating cardiopulmonary resuscitation. Soon after return of spontaneous circulation was achieved, he once again experienced pulseless electrical activity arrest, was resuscitated once more, and subsequently intubated. A repeat CXR done showed a widened mediastinum (Fig. 4). A chest CT without contrast was obtained demonstrating an 8 cm dilatation of the aortic root.
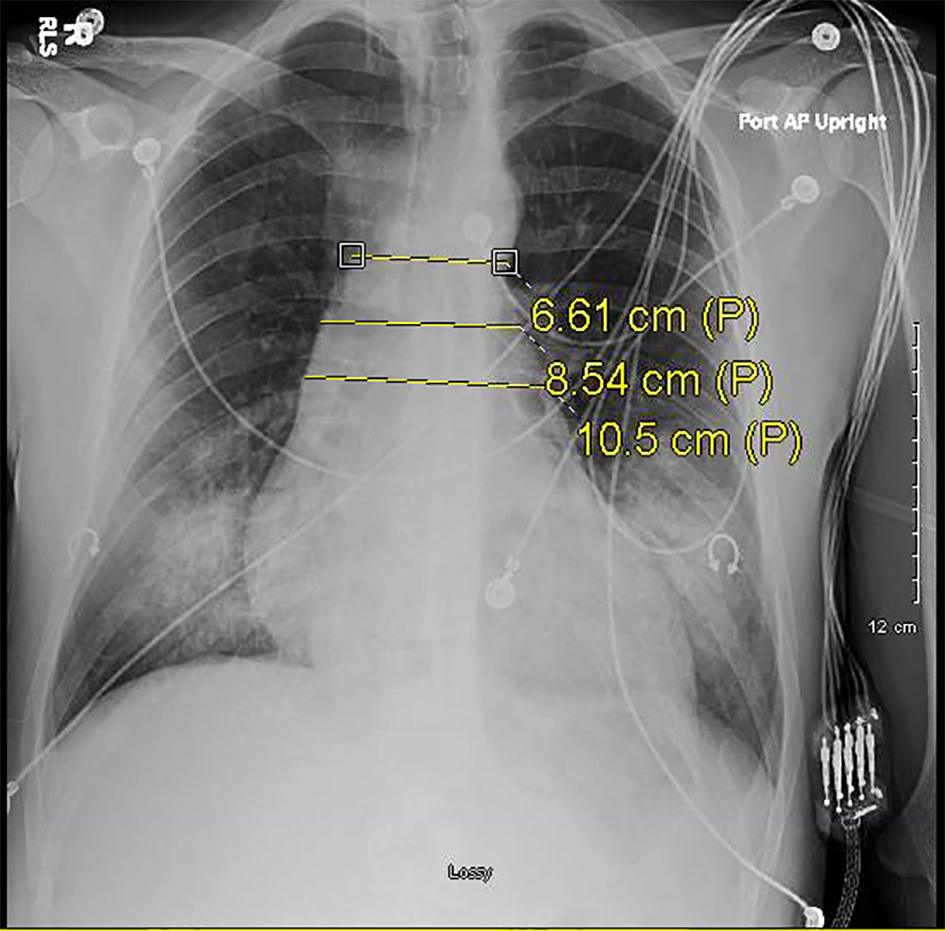 Click for large image | Figure 3. Serial plain radiograph images demonstrating progressively widening mediastinum on December 3, 2016. |
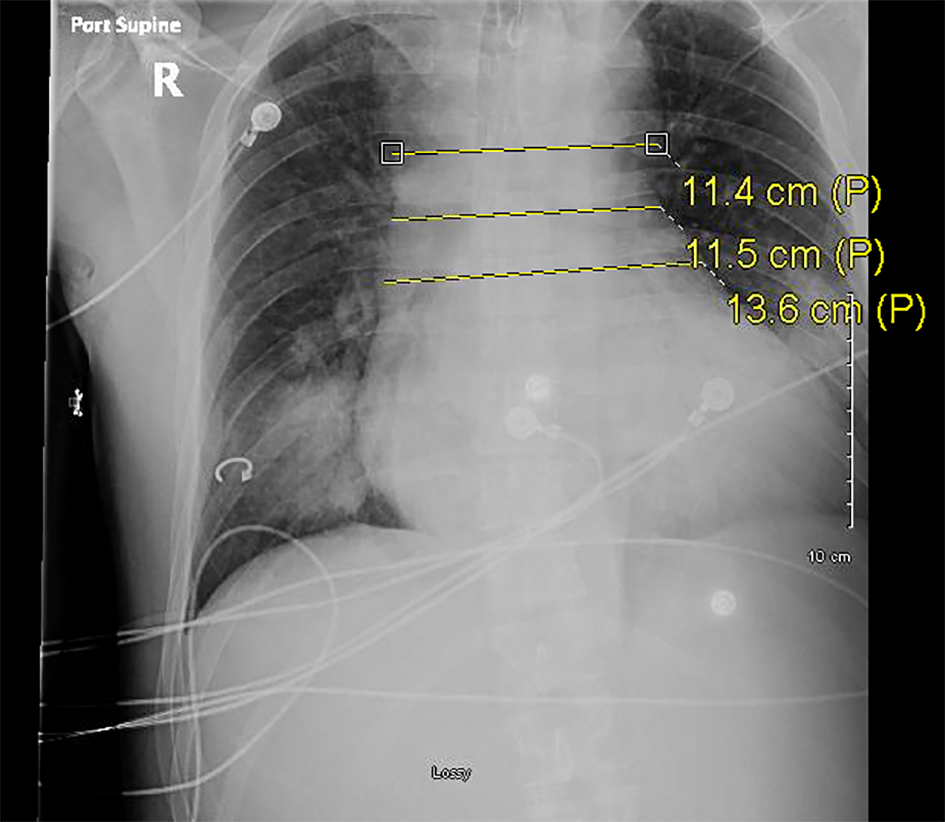 Click for large image | Figure 4. Serial plain radiograph images demonstrating progressively widening mediastinum on December 4, 2016. |
A chest CT with contrast revealed a focal point of dissection in the ascending aorta and aortic insufficiency (Fig. 5). The patient was taken emergently to the operating room (OR). A Bentall procedure involving aortic root, valve, and ascending aorta graft replacement with re-implantation of the coronary arteries into the graft was performed. The procedure was complicated by cardiac tamponade leading to hematoma formation which required the patient to return to the OR for evacuation. After evacuation, the patient was returned to the SICU in stable condition without inotropic support. This patient’s hospital course was also complicated by acute kidney injury requiring hemodialysis, ischemic hepatitis, and anoxic brain injury all secondary to cardiogenic shock. Over the course of the next 3 weeks, the patient’s kidney function recovered and he no longer needed dialysis. His liver function tests also returned to normal. Neurologically, he regained some function and was subsequently discharged to subacute rehabilitation.
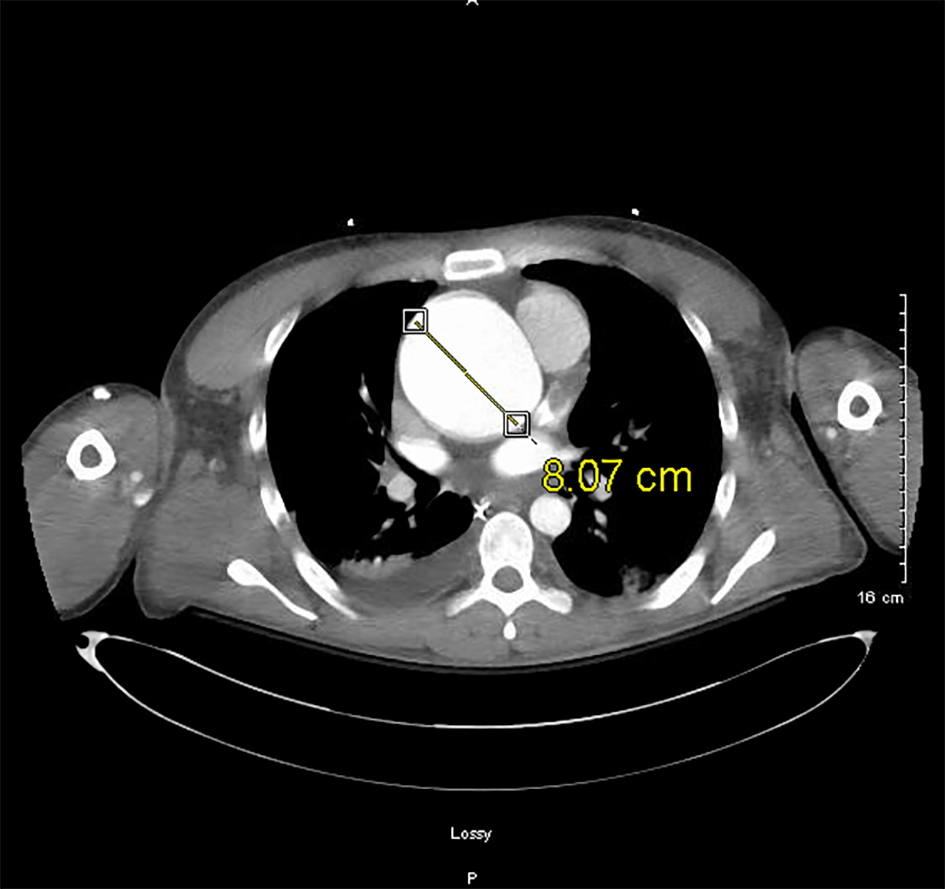 Click for large image | Figure 5. CTA of the chest demonstrating 8 cm aortic root dilatation on December 5, 2016. |
In this patient, a diagnosis of Marfan syndrome was made clinically based on his presentation. At the time of discharge, a definitive genetic diagnosis was not yet performed. Survival of his cardiac events can be likely attributed to his left dominant coronary circulation, a trait found in only 18% of the population.
| Discussion | ▴Top |
Marfan Syndrome (MFS) is one of the most common connective tissue diseases, with a reported prevalence of 1 in 3,000 to 5,000 individuals [1]. It is primarily of autosomal dominant inheritance, with 90% of cases involving a mutation in the gene for connective tissue protein, FBN1. The other 10% are with a complete absence of the gene altogether [2]. The wide range in prevalence reported is largely due to the widely variable phenotypic expression of symptoms in Marfan syndrome. Classically, clinicians view the disorder as the triad of musculoskeletal, ocular, and cardiac abnormalities, including aortic root dilatation and ectopia lentis [3]. The revised Ghent criteria, developed in 2010, aim to systematically diagnose Marfan patients because the overlap of normal phenotypic variability and Marfan syndrome is so great [4]. Our patient, as described earlier, meets the diagnosis of Marfan syndrome based on the major criteria of aortic criterion, and ≥ 7 systematic score (see modified Ghent criteria for more information) [5].
Aortic root pathology is the most common cause of morbidity and mortality in patients with MFS, and includes aneurysmal dilation, and aortic regurgitation and dissection. Undiagnosed MFS is often associated with aortic dissection with a frequency is as high as 50% in patients under the age of 40 [6]. Likely, the initial findings of dyspnea, tachycardia, and chest pain with inspiration in this patient, are a result of compression of the tracheobronchial tree from an expanding thoracic aortic aneurysm [4]. Initial laboratory results are often non-yielding in thoracic aortic aneurysm, as in this case; however, vital signs can be helpful but non-specific. Sinus tachycardia is the most common initial change in vital signs, and changes in distal pulses may be noted [7]. The condition of this patient became guarded after presumably beginning to dissect the already damaged thoracic aorta. As evident from the images, the mediastinum continued to widen considerably. Acute hemodynamic compromise is the likely precipitating factor for the series of initial cardiac arrests, although it can easily be explained by acute myocardial ischemia due to coronary occlusion. When the dissection reaches the coronary arteries, the right is usually affected, which can lead to complete heart block, infarction, and death [8]. An estimated 80% of the population has right coronary dominance [9]. Fortunately for this patient, his coronary anatomy is such that it is left-dominant so the intrinsic pacemaker of the heart remained perfused. Left coronary dominance is only seen in approximately 18% of the population [9].
Surgical management is the mainstay of treatment in aortic dissection, which was done in this patient to reduce the already high rate of mortality [6]. Although the surgery was complicated by hematoma formation, the risks and benefits of surgery still outweighed the alternative of medical management. Prevention in this patient should have been aimed at making diagnosis of Marfan syndrome long before aortic pathologies arose. Next steps include testing relatives for Marfan syndrome due to the autosomal dominant nature of the disease, as well as preventing future pathologies in this patient by controlling blood pressures and close follow-up.
| References | ▴Top |
- Judge DP, Dietz HC. Marfan’s syndrome. Lancet. 2005;366(9501):1965-1976.
doi - Hilhorst-Hofstee Y, Rijlaarsdam ME, Scholte AJ, Swart-van den Berg M, Versteegh MI, van der Schoot-van Velzen I, Schabitz HJ, et al. The clinical spectrum of missense mutations of the first aspartic acid of cbEGF-like domains in fibrillin-1 including a recessive family. Hum Mutat. 2010;31(12):E1915-1927.
doi pubmed - Dietz HC (Updated June 30, 2009). Marfan syndrome. In: Gene Reviews at Gene Test: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1997-2011. Available at http://www.genetests.org.
- Radonic T, de Witte P, Groenink M, de Bruin-Bon RA, Timmermans J, Scholte AJ, van den Berg MP, et al. Critical appraisal of the revised Ghent criteria for diagnosis of Marfan syndrome. Clin Genet. 2011;80(4):346-353.
doi pubmed - Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47(7):476-485.
doi pubmed - Mehta RH, O’Gara PT, Bossone E, Nienaber CA, Myrmel T, Cooper JV, Smith DE, et al. Acute type A aortic dissection in the elderly: clinical characteristics, management, and outcomes in the current era. J Am Coll Cardiol. 2002;40(4):685-692.
doi - Bicknell CD, Powell JT. Thoracic aortic aneurysms. Br J Surg. 2013;100(7):850-852.
doi pubmed - Bossone E, et al. Coronary artery involvement in patients with acute type A aortic dissection: Clinical characteristics and in-hospital outcomes. J Am Coll Card. 2003;41S:235.
doi - Das Hirak, et al. A study of coronary dominance in the population of Assam. Journal of Anatomical Society of India. 2010;59(2):187-191.
doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.