

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 9, Number 3, March 2018, pages 103-105
Management of an Unknown Primary Merkel Cell Carcinoma: A Case Report
Aikaterini Papadopouloua, Pinelopi Theopisti Memtsaa, b, Antonio Capizzelloa, Kyriaki Pisteuou Gompakia
aDepartment of Radiation Oncology, Aristotle University of Thessaloniki, Greece
bCorresponding Author: Pinelopi Theopisti Memtsa, Department of Radiation Oncology, Aristotle University of Thessaloniki, Psarron 26, 54453, Thessaloniki, Greece
Manuscript submitted January 12, 2018, accepted January 29, 2018
Short title: Unknown Primary Merkel Cell Carcinoma
doi: https://doi.org/10.14740/jmc3004w
| Abstract | ▴Top |
Merkel cell carcinoma (MCC) represents a rare skin cancer associated with sun exposure. It is an uncommon skin neoplasm of the elderly population. The majority of patients with MCC present with localized disease at diagnosis and few patients have regional lymph node (LN) involvement and distant metastases. Although it has been previously reported in various anatomical sites, LN metastatic MCC in the absence of a primary site is extremely rare and for this reason there is no standard approach to its management. Currently, there is no standard approach to the management of MCC in the absence of a primary site. Since the disease is highly aggressive and the failure rate following surgery alone is high, radiotherapy (RT) is usually administered as temporary support for numerous patients with MCC.
Keywords: Radiotherapy; Merkel cell carcinoma; Unknown primary
| Introduction | ▴Top |
Merkel cell carcinoma (MCC) represents a rare skin cancer associated with sun exposure affecting mainly Caucasian males over the sixth decade. Although the origin of Merkel cell has been debated, MCC is considered a neuroendocrine tumor (NET) due to its characteristic histopathologic appearance expressing CD56, neurofilament protein (NFP) and chromogranin A [1].
It was first described by Toker in 1972 [2]. It is an uncommon skin neoplasm of the elderly population. The majority of patients with MCC present with localized disease at diagnosis and few patients have regional lymph node (LN) involvement and distant metastases. Although it has been previously reported in various anatomical sites, LN metastatic MCC in the absence of a primary site is extremely rare and for this reason there is no standard approach to its management [3].
Curative surgery is commonly recommended to manage localized MCC. Nevertheless, specific postoperative palliative treatments for MCC have emerged, including radiotherapy (RT) or chemotherapy, due to the high local failure rate and the aggressive nature of the disease [4].
We present a case of a 63-year-old patient who underwent a diagnostic biopsy of an inguinal lump at his local hospital which was proven to be an infiltrated LN by an MCC. Following his imaging staging with computed tomography (CT) and positron emission tomography (PET) scan, which showed only abdominal LN disease, she was referred to our department for adjuvant RT after surgery and chemotherapy.
| Case Report | ▴Top |
A 63-year-old female was admitted to her local hospital with edema of the left lower extremity and motor dysfunction. On clinical examination, a large palpable inguinal mass was identified. It was oval, lobulated, firm, fixed to underlying tissue with normal overlying skin. She reported a history of a palpable mass measuring 2 cm of 2 years’ duration in the left inguinal region with progressive growth. The patient’s medical history was not significant and no suspicious skin lesions were identified. Triplex US of the left extremity revealed normal depiction of the deep veins of the lower extremity without thrombosis but in the abdominal region, swollen rounded LNs were observed. Diagnostic workup imaging including abdominal-pelvic contrast-enhanced CT scan revealed a large lobulated mass measuring 7 × 7 cm in the left inguinal area adjacent to the femoral vessels and an enlarged left external iliac LN of size 3.7 × 3 cm. The mass was compressing the left common femoral vein and remained inseparable from the vein as well as from the adductor muscles ventrally. Further diagnostic evaluation including CT of the chest and abdomen demonstrated only a cyst in the right kidney. The results of laboratory examinations, including complete blood count, renal, bone hepatic and coagulation profiles, carcinoembryonic antigen (CEA), alfa-fetoprotein (AFP) and CA-12-5, revealed no abnormalities. With no primary site of cancer identified, fine-needle aspiration cytology (FNAC) of the mass was performed. According to the histological report, it was a malignancy with extensive necrosis, but it did not result in a definitive diagnosis. The differential diagnosis included malignant neoplasm of small cells and lymphoma. Thus, the patient underwent an excisional biopsy of the left inguinal lymph node. The biopsy specimen showed a low-grade neoplasm with morphological and immunohistochemical characteristics, which were consistent with the diagnosis of MCC. Histopathologically, the neoplastic cells were monomorphic, basophilic, small, round with ovoid vesicular nuclei and scanty cytoplasm. Immunohistochemically, the tumor cells were positive for LMWKer, Ker20, Nf, synaptophysin and chromogranin. The tumor cells were negative for TTF1, Ker7, HMWKer, p63, Ker5/6, S-100, c-Kit and LCA. Because of the size and involvement of the femoral vessels, chemotherapy was recommended as the most appropriate curative option by the multidisciplinary team. Systemic chemotherapy was administered as six cycles cisplatin/etoposide- based combination schedule with remarkable clinical response. After chemotherapy, the CT imaging study revealed a reduction in size of the inguinal LN measuring 3.6 × 2.6 cm. The patient refused further treatment. Nonetheless, she was admitted again to the hospital 5 months later due to disease progression. The CT demonstrated the presence of the known lymphadenopathy in the left inguinal area with increased dimensions. She managed with partial left inguinal LN surgical excision due to invasion of the safenofemoral junction. The removal part was 14 × 9 × 3 cm and the histological examination showed almost complete invasion of the known malignant cells according to the patient’s history of MCC. Post-surgical PET scan showed low tracer uptake in the left external iliac node (SUVmax: 3.1) and in the left inguinal area (SUVmax: 3.5), with possible reactive etiology. Pathological tracer uptake of 18F-FDG (SUVmax: 3.4) was observed in the left adrenal gland, a finding that could be related to adenoma. There was no evidence of metabolically active lesions in the skin or internal organs. Subsequently, she attended to our Radiotherapy Department after 3 weeks so as to be delivered to her adjuvant radiation therapy. She received a total dose of 60 Gy in 30 fractions with a daily dose of 2 Gy in the inguinal region and a total dose of 46 Gy in the external iliac lymph nodes (Fig. 1).
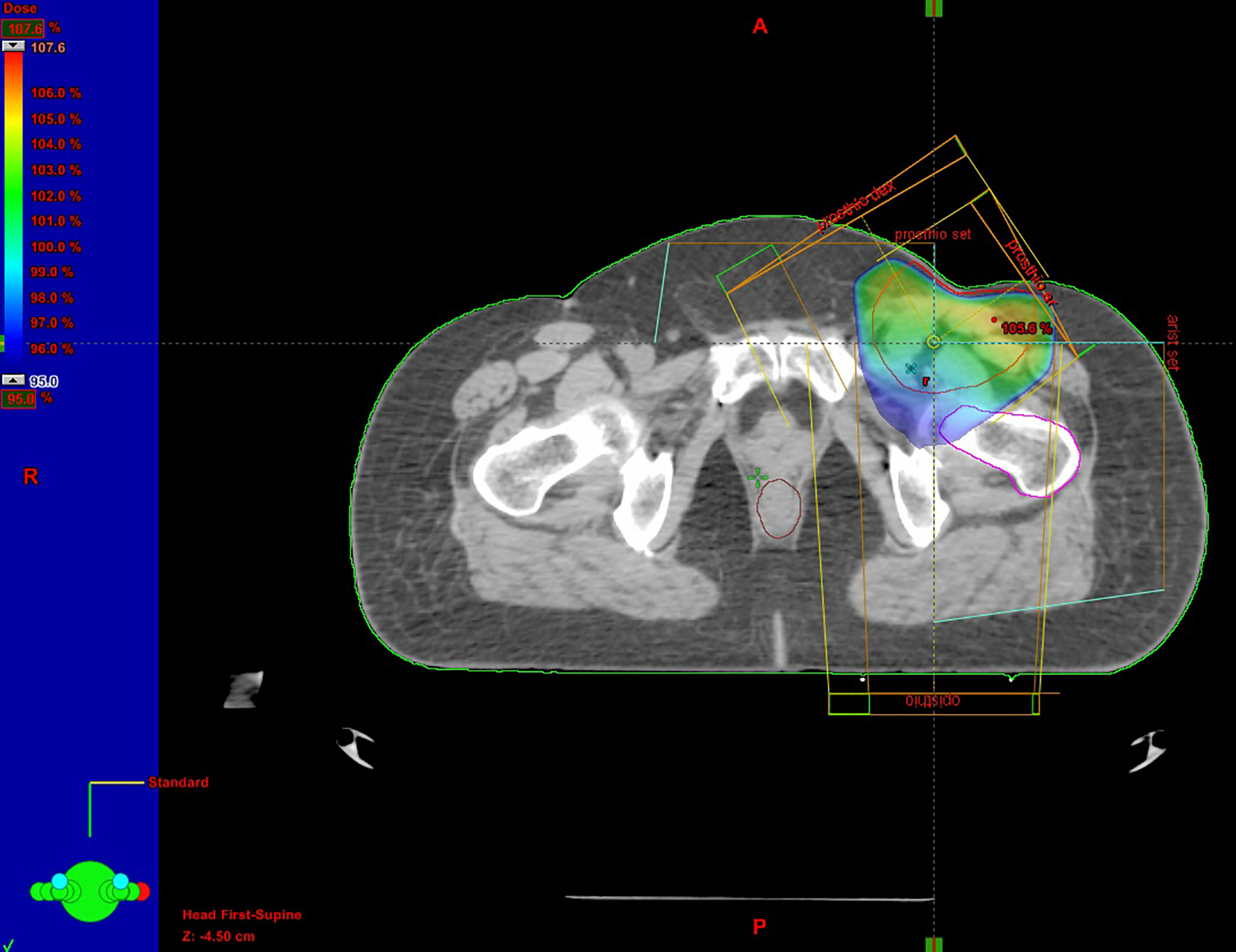 Click for large image | Figure 1. Radiotherapy treatment plan. |
| Discussion | ▴Top |
It is difficult to diagnose MCC in an LN due to its similarity to other poorly differentiated small basophilic cell tumors, including metastatic melanoma, lymphoma, small cell carcinoma, and neuroendocrine carcinoma metastatic from other organs. Immunohistochemistry is necessary for the definitive diagnosis. On immunohistochemical staining, it is necessary to express the pattern of CK20 positivity along with positivity for any of the neuroendocrine markers such as, chromogranin A, synaptophysin or neuron specific enolase (NSE) and negative TTF-1 staining [3].
The origin of MCC has been debated and both neural crest cells as well as epithelial cells have been implicated. Due to the rarity of this malignancy, it has not yet been delineated in the case of metastatic to LNs unknown primary Mercel cell carcinoma (UPMCC), if the tumor arises de novo from neural cells located within the involved LNs or if the primary lesion undergoes spontaneous regression. It has been shown in MCC of a known skin primary there is clear association of the carcinogenesis process with infection from polyomavirus which is on the contrary is not seen in cases of UPMCC [5].
A chest, abdomen and pelvis CT scan usually confirms LN involvement and reveals the extent of disease. Recently the role of PET/CT has been upgraded in depicting the sites of LN disease in MCC due to its high reported specificity and sensitivity, 98% and 90% respectively [6].
Currently, there is no standard approach to the management of MCC in the absence of a primary site. Mohs micrographic surgery is currently considered as the primary and complementary measure for controlling this serious disease. Since the disease is highly aggressive and the failure rate following surgery alone is high, RT is usually administered as temporary support for numerous patients with MCC. MCC cell lines have been demonstrated to be radiosensitive in vitro. Results have indicated that adjuvant RT, following initial surgery and resection for recurrent MCC and palliation is beneficial. A previous study reported that a radiation dose of 45 Gy had significant impact on local control and prolonged survival in nine patients, whereas a subset of seven patients who received < 45 Gy had a poorer outcome. MCC is considered to be resistant to chemotherapy; however, various agents have been used to treat MCC with variable results. The most commonly used chemotherapy regimen is etoposide/cisplatin [7].
Although multimodal treatment with surgery, RT and chemotherapy results in excellent local control, local recurrence and distant metastases are ultimately developed, possibly owing to the characteristics of MCC or inadequate treatment. Therefore, the role of adjuvant postoperative chemotherapy and RT in MCC remains to be determined in future trials.
In conclusion, MCC presenting with LN involvement without an obvious primary site is uncommon and the diagnosis can be challenging. Meticulous workup for the primary focus as well as thorough pathological and immunohistochemical analysis must be carried out. Advances in understanding the origin and natural history of this rare entity would be helpful in establishing management guidelines in the future. Further investigations are needed to delineate the true natural history of this rare disease as well as the single best approach to treatment [8].
| References | ▴Top |
- Agelli M, Clegg LX. Epidemiology of primary Merkel cell carcinoma in the United States. J Am Acad Dermatol. 2003;49(5):832-841.
doi - Toker C. Trabecular carcinoma of the skin. Arch Dermatol. 1972;105(1):107-110.
doi pubmed - Poulsen M. Merkel-cell carcinoma of the skin. Lancet Oncol. 2004;5(10):593-599.
doi - Kotteas EA, Pavlidis N. Neuroendocrine Merkel cell nodal carcinoma of unknown primary site: management and outcomes of a rare entity. Crit Rev Oncol Hematol. 2015;94(1):116-121.
doi pubmed - Deneve JL, Messina JL, Marzban SS, Gonzalez RJ, Walls BM, Fisher KJ, Chen YA, et al. Merkel cell carcinoma of unknown primary origin. Ann Surg Oncol. 2012;19(7):2360-2366.
doi pubmed - Treglia G, Kakhki VR, Giovanella L, Sadeghi R. Diagnostic performance of fluorine-18-fluorodeoxyglucose positron emission tomography in patients with Merkel cell carcinoma: a systematic review and meta-analysis. Am J Clin Dermatol. 2013;14(6):437-447.
doi pubmed - Ott MJ, Tanabe KK, Gadd MA, Stark P, Smith BL, Finkelstein DM, Souba WW. Multimodality management of Merkel cell carcinoma. Arch Surg. 1999;134(4):388-392; discussion 392-383.
- Tseng J, Dhungel B, Mills JK, Diggs BS, Weerasinghe R, Fortino J, Vetto JT. Merkel cell carcinoma: what makes a difference? Am J Surg. 2015;209(2):342-346.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.