

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 9, Number 7, July 2018, pages 229-232
Bloodless Glomeruli: A Case Report on Scleroderma Renal Crisis
Sunit Toliaa, c, Hassan Kassemb, Samira Ahsana
aDepartment of Internal Medicine, Providence-Providence Park Hospital/Michigan State University College of Human Medicine, 16001 W. Nine Mile Road, Southfield, MI 48075, USA
bDepartment of Medical Education, Providence-Providence Park Hospital/Michigan State University College of Human Medicine, 16001 W. Nine Mile Road, Southfield, MI 48075, USA
cCorresponding Author: Sunit Tolia, 16001 W. Nine Mile Road, Southfield, MI 48075, USA
Manuscript submitted February 5, 2018, accepted February 13, 2018
Short title: Bloodless Glomeruli
doi: https://doi.org/10.14740/jmc3018w
| Abstract | ▴Top |
Scleroderma renal crisis (SRC) is a main complication of systemic sclerosis affecting approximately one-fifth of the patient population with the disease. We present a case of a 38-year-old African American female who initially presented with findings suggestive of thrombotic thrombocytopenic purpura; however, the clinical manifestations, laboratory findings and renal biopsy confirmed the diagnosis of SRC. The case is worth reporting in medical literature because of its unique presentation and to note the findings of the kidney biopsy. SRC can present initially as thrombotic thrombocytopenic purpura; therefore, clinicians need to be more aware of this entity and it should be considered in the differential diagnosis of microangiopathic hemolytic anemia. This case also illustrates that the classic “onion skin appearance” on kidney biopsy in SRC is not always present and that patients can have “bloodless” glomeruli.
Keywords: Scleroderma; Scleroderma renal crisis; Microangiopathic hemolytic anemia; Thrombocytopenia; Thrombotic thrombocytopenic purpura
| Introduction | ▴Top |
Scleroderma is a heterogeneous autoimmune condition of unknown etiology that is divided into localized and systemic (SSc) classifications. SSc is subdivided into limited (lcSSc) and diffuse (dcSSc) forms based on the amount of skin tightening and visceral organ involvement. The estimated incidence of SSc is approximately 20 cases per million per year and scleroderma renal crisis (SRC) affects about 5-10% of those patients [1]. The manifestations of SRC include new onset hypertension, microangiopathy, deteriorating renal function and neurological symptoms. Those with dcSSc are at a 25% risk of developing SRC, as compared to only 1% risk in patients who have lcSSc [1].
The differential diagnosis of microangiopathic hemolytic anemia (MAHA) is SRC, thrombotic thrombocytopenic purpura (TTP), hemolytic uremic syndrome (HUS) and malignant hypertension. Clinically, it is difficult to distinguish TTP from SRC due to similar renal and neurological features. TTP presents with thrombocytopenia, purpura, rapidly progressive renal failure and absence or marked reduction in a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS-13) activity. However, since these conditions are life-threatening diseases, differentiating between them is crucial to the survival of the patient.
| Case Report | ▴Top |
A 38-year-old African American woman with a history of systemic lupus erythematous (SLE) presented with a chief complaint of “difficulty breathing, vomiting and diarrhea”. According to the husband, she has been suffering from increased number of SLE flare-ups in the past year since the onset of her diagnosis. The patient’s treatment regimen included hydroxychloroquine 200 mg twice daily and was recently started on oral steroid medication.
On the initial assessment, the patient’s blood pressure was 139/50 mm Hg, heart rate of 113 beats/min and respiratory rate of 40 breaths/min. She was toxic in appearance and unresponsive. Therefore, she was emergently intubated due to worsening primary disorder of metabolic acidosis and concomitant respiratory acidosis. Physical examination revealed dry mucous membranes, bilateral crackles and rhonchi in all lung fields and no skin changes over her extremities. Chest X-ray findings showed extensive diffuse bilateral airspace disease.
In addition, the patient required emergent hemodialysis for worsening acidosis, hyperkalemia (6.4 mmol/L), elevated blood urea nitrogen (BUN) level (103 mg/dL), elevated creatinine level (19.2 mg/dL), glomerular filtration rate (GFRA) of 2.48 and progressive oliguria. The urine analysis showed proteinuria (500 mg/dL), hematuria and waxy casts. Laboratory analysis revealed the following values: white blood cell count 15.0/µL, hemoglobin 7.2 g/dL, platelet count 128,000/µL, absolute reticulocyte count 93.9 K/mcL, reticulocytes 3.07%, direct bilirubin 0.5 mg/dL, total bilirubin 0.8 mg/dL, lactate dehydrogenase (LDH) 1,097, haptoglobin < 10 mg/dL and complement C3 levels of 46 mg/dL and C4 levels of 18 mg/dL. Furthermore, the complete blood count (CBC) correlated with findings of MAHA and thrombocytopenia with a peripheral smear showing schistocytes (Fig. 1).
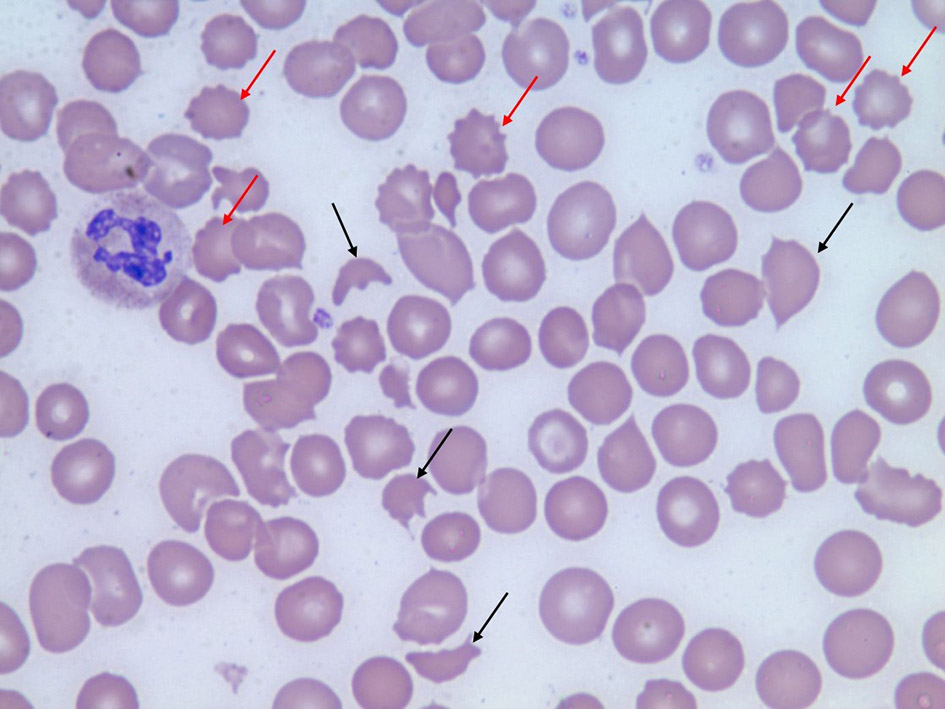 Click for large image | Figure 1. Peripheral blood smear shows multiple schistocytes (black arrows) and burr cells (red arrows). |
Based on the clinical presentation, the differential diagnosis was TTP, HUS, SRC, lupus nephritis and malignant hypertension. Upon admission, the patient was empirically started on plasmapheresis therapy for treatment of TTP/HUS and a ADAMTS-13 level was ordered. Plasmapheresis was discontinued on day 7 because ADAMTS-13 level was within normal range. On day 3, her blood pressure began to rise with systolic blood pressure ranging from 160 to 180 mm Hg and diastolic blood pressure ranging from 80 to 130 mm Hg. She was started on captopril for the treatment of hypertension. Furthermore, her anti-scleroderma-70 (anti-Scl-70) antibody titers were greater than 8.0 AI (normal range: 0 - 0.9 AI). Her anti-nuclear antibody (ANA) titers were positive at 320 TU in a homogeneous pattern and a titer of 320 TU in a nucleolar pattern. Additional immunological studies and infectious titers were ordered (Table 1).
 Click to view | Table 1. Following Diagnostic Tests Were Ordered and the More Relevant Results Are Highlighted in Italic |
The complement levels were within normal range and lupus nephritis was ruled out of the differential diagnosis. Based on the worsening renal function and immunological workup suggestive of scleroderma, SRC was not ruled out.
Once she was more hemodynamically stable, a renal biopsy was ordered. The results showed “bloodless” appearing glomeruli, a pattern of morphological changes typically seen in thrombotic microangiopathy (Fig. 2 & 3). As mentioned, the differential diagnosis of thrombotic microangiopathy includes: TTP, HUS, SRC and malignant hypertension. Prior diagnostic studies such as ADAMTS-13 gene activity and kidney biopsy ruled out TTP and HUS. Based on the development of new-onset of oliguric acute renal injury, accelerated hypertension, positive anti-Scl-70 titers, lab findings suggestive of MAHA with schistocytes on peripheral blood smear and kidney biopsy results, the patient was diagnosed with SRC.
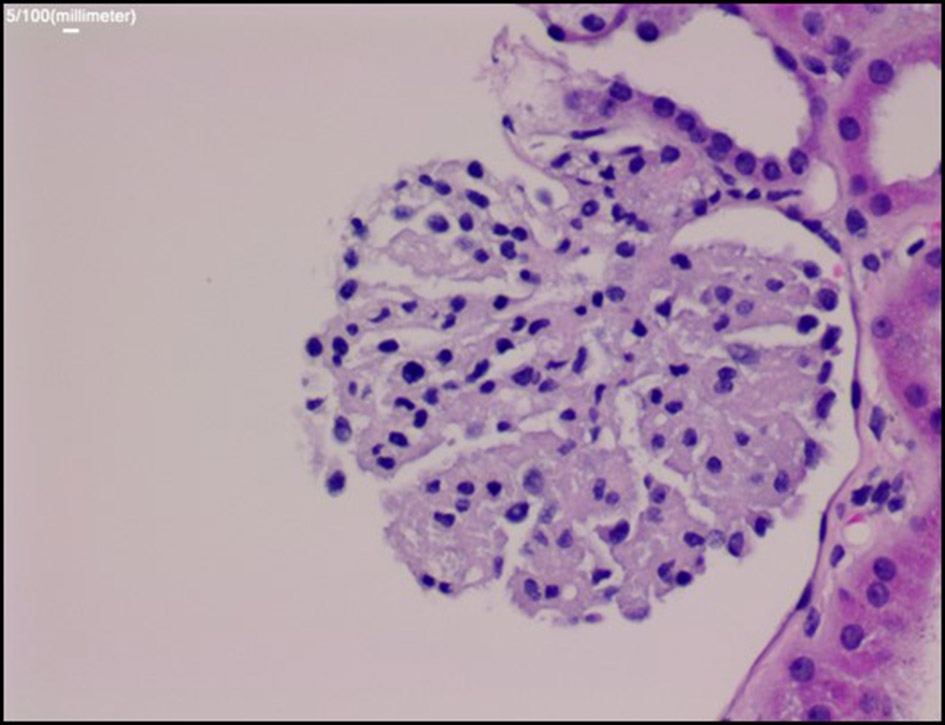 Click for large image | Figure 2. Periodic acid-Schiff stain: the glomerulus demonstrates global collapse of glomerular capillaries with “bloodless” appearance. |
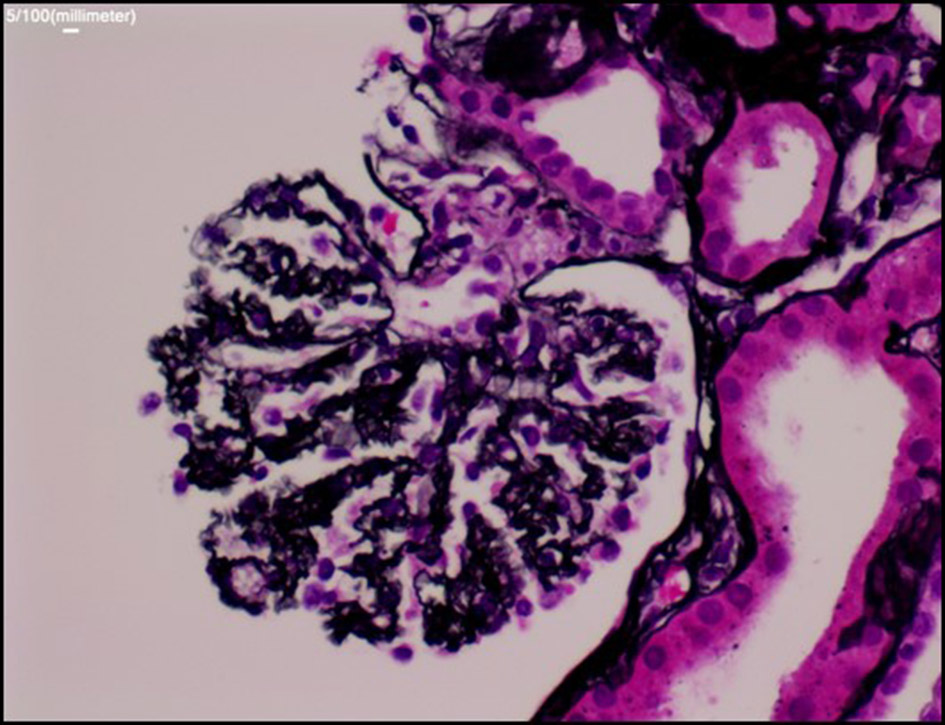 Click for large image | Figure 3. Silver stain: the glomerular basement membrane (GBM) is diffusely wrinkled and collapsed, findings similar to ischemic changes. But no microthrombi or widening of subendothelial space are seen in the glomerular capillaries. There are no crescent or proliferative changes. |
| Discussion | ▴Top |
Scleroderma is a systemic disease characterized by autoimmune activation, endothelial dysfunction and vascular damage [2]. Localized scleroderma is the most common form of the disease and manifests with cutaneous involvement without any organ dysfunction. The incidence of the disease is 0.3 - 3.0 per 100,000 cases per year and it primarily affects children [3]. The etiology is unknown, but it is fairly a benign and self-limiting condition. It is subdivided into five different categories: linear scleroderma, plaque morphea, deep morphea, bullous morphea and generalized morphea. The term morphea is characterized by skin thickening due to increased qualities of collagen in the indurative lesion [4]. After thorough literature review, many studies have shown that local scleroderma is not only confined to cutaneous disease. According to Christen-Zaech et al, about 20% of patients develop extracutaneous manifestations such as arthritis, uveitis, seizures and restrictive lung conditions [5, 6]. Neurological involvement has been shown to be the most common, specifically complex partial seizures [7].
Similarly SSc is defined as a chronic connective tissue disorder with cutaneous findings along with severe visceral organ involvement. This condition is due to endothelial cell injury, vascular hyperreactivity, obliterative microvasculopathy and excessive collagen deposition. It is further subdivided into lcSSc and dcSSc forms due to the amount of skin tightening [8]. The lcSSc disease formerly known as CREST syndrome (calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly and telangiectasias), involves skin tightening distally to the elbows and knees. On the contrary, dcSSc involves skin tightening of the proximal extremities and trunk and carries a higher risk of visceral involvement.
Among the many severe complications of dcSSc, SRC occurs in about 5-10% of patients with SSc [1]. In a large retrospective analysis by Penn et al, 110 SRC cases were reviewed between 1990 and 2005 and he concluded that 12% of dcSSc patients and 2% of lcSSc patients developed SRC [9]. SRC is defined as new onset accelerated hypertension (> 150/85 mm Hg) or rapidly progressive oliguric/anuric acute renal failure in the setting of SSc. The manifestations of this condition include a decline in estimated GFR (eGFR) > 30%, thrombocytopenia, MAHA, hematuria and proteinuria [10]. Our patient presented with all of the following clinical features.
According to Shanmugam et al, normotensive SRC is linked with a worse outcome than hypertensive SRC [11]. On day 3 of admission, our patient presented with new onset of elevated blood pressure, prompting us to begin angiotensin-converting-enzyme inhibitor (ACEi) therapy. Captopril is the first-line treatment for SRC due to its short half-life and easy titration. The goal of therapy is to lower the systolic blood pressure down by 20 mm Hg and diastolic blood pressure down by 10 mm Hg in a 24-h period. The administration of early ACEi therapy has decreased SRC mortality at 1 year from 76% to 15% [12]. Among patients who are undergoing hemodialysis, about 30% regain renal functional recovery while continuing ACEi with dialysis [12]. However, even though ACEi has shown promising results, the overall 5-year mortality rate worldwide remains between 30% and 50% [9, 13].
Some risk factors of SRC to note are progressive skin disease, high-dose corticosteroid therapy of > 15 mg/day in the preceding 6 months, presence of antibodies to RNA polymerase, anemia, joint contractures, tendon friction rubs, pericardial effusion and cardiac insufficiency [14]. Renal biopsy is one of the key elements of making the diagnosis of SRC [15]. However, the three glomeruli that were obtained from the patient’s renal medulla demonstrated a “bloodless” appearance. Given the results of “bloodless” glomeruli on renal biopsy, this presents a unique finding in patients with SRC. Typical renal biopsy results include “onion skinning” representative of fibrointimal proliferation with near obliteration of the vascular lumen and glomeruli ischemia [10]. This case is unique in the terms of how the initial presentation was suggestive of TTP, but based on the clinical presentation, risk factors, laboratory findings, and renal biopsy results, the underlying diagnosis was SRC. Furthermore, the kidney biopsy did not show the classic “onion skinning” appearance, instead the renal glomeruli were bloodless.
Conclusion
SRC is one of the most feared complications of SSc due to its high level of mortality. This case report describes a rare presentation of a patient whose labs were initially concerning for TTP and acute renal failure; however, the underlying etiology was SRC. In addition, the case had a unique pathological finding of “bloodless” glomeruli, whereas patients usually have a concentric “onion skin” narrowing of arterioles and glomerular ischemia. It is clinically difficult to differentiate between MAHA conditions due to similar renal and neurological manifestations. Therefore, clinicians must conduct a thorough clinical workup to begin prompt aggressive therapy that will result in an optimal outcome.
Acknowledgments
We would like to acknowledge April Nofzinger D.O. from the department of hematology and oncology at our institution for providing the peripheral smear.
Conflict of Interest
The authors declare that there is no conflict of interests regarding the publication of this paper.
| References | ▴Top |
- Nikpour M, Stevens WM, Herrick AL, Proudman SM. Epidemiology of systemic sclerosis. Best Pract Res Clin Rheumatol. 2010;24(6):857-869.
doi pubmed - Kurasawa K, Hirose K, Sano H, Endo H, Shinkai H, Nawata Y, Takabayashi K, et al. Increased interleukin-17 production in patients with systemic sclerosis. Arthritis Rheum. 2000;43(11):2455-2463.
doi - Peterson LS, Nelson AM, Su WP, Mason T, O'Fallon WM, Gabriel SE. The epidemiology of morphea (localized scleroderma) in Olmsted County 1960-1993. J Rheumatol. 1997;24(1):73-80.
pubmed - Matsuura K, Umebayashi Y, Otsuka F. Computed tomography reveals thickened subcutaneous tissue in scleroedema. Br J Dermatol. 1997;137(6):1015-1016.
doi pubmed - Christen-Zaech S, Hakim MD, Afsar FS, Paller AS. Pediatric morphea (localized scleroderma): review of 136 patients. J Am Acad Dermatol. 2008;59(3):385-396.
doi pubmed - Zulian F, Vallongo C, Woo P, Russo R, Ruperto N, Harper J, Espada G, et al. Localized scleroderma in childhood is not just a skin disease. Arthritis Rheum. 2005;52(9):2873-2881.
doi pubmed - Appenzeller S, Montenegro MA, Dertkigil SS, Sampaio-Barros PD, Marques-Neto JF, Samara AM, Andermann F, et al. Neuroimaging findings in scleroderma en coup de sabre. Neurology. 2004;62(9):1585-1589.
doi pubmed - LeRoy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA, Jr., Rowell N, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;15(2):202-205.
pubmed - Penn H, Howie AJ, Kingdon EJ, Bunn CC, Stratton RJ, Black CM, Burns A, et al. Scleroderma renal crisis: patient characteristics and long-term outcomes. QJM. 2007;100(8):485-494.
doi pubmed - Toescu SM, Mansell A, Dinneen E, Persey M. Steroid-induced scleroderma renal crisis in an at-risk patient. BMJ Case Rep. 2014;2014.
- Shanmugam VK, Steen VD. Renal disease in scleroderma: an update on evaluation, risk stratification, pathogenesis and management. Curr Opin Rheumatol. 2012;24(6):669-676.
doi pubmed - Steen VD. Scleroderma renal crisis. Rheum Dis Clin North Am. 2003;29(2):315-333.
doi - Teixeira L, Mouthon L, Mahr A, Berezne A, Agard C, Mehrenberger M, Noel LH, et al. Mortality and risk factors of scleroderma renal crisis: a French retrospective study of 50 patients. Ann Rheum Dis. 2008;67(1):110-116.
doi pubmed - DeMarco PJ, Weisman MH, Seibold JR, Furst DE, Wong WK, Hurwitz EL, Mayes M, et al. Predictors and outcomes of scleroderma renal crisis: the high-dose versus low-dose D-penicillamine in early diffuse systemic sclerosis trial. Arthritis Rheum. 2002;46(11):2983-2989.
doi pubmed - Hassan AT, Elbadry MI, Tawfik MA. A case report of thrombotic thrombocytopenic purpura associated with systemic lupus erythematosus: overlapping features. American Journal of Medical Case Reports 2014;2:206-213.
doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.