

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 9, Number 10, October 2018, pages 332-337
Acute Acalculous Cholecystitis: A Rare Presentation of Autoimmune Hepatitis and Primary Sclerosing Cholangitis Overlap Syndrome
Feng Yina, Peter V. Draganovb, Jinping Laia, Steven J. Hughesc, Roberto J. Firpi-Morellb, Xiuli Liua, d
aDepartment of Pathology, Immunology and Laboratory Medicine, College of Medicine, University of Florida, Gainesville, FL, USA
bDepartment of Gastroenterology, Hepatology and Nutrition, College of Medicine, University of Florida, Gainesville, FL, USA
cDepartment of Surgery, College of Medicine, University of Florida, Gainesville, FL, USA
dCorresponding Author: Xiuli Liu, Department of Pathology, Immunology and Laboratory Medicine, P.O. Box 100275, Gainesville, FL 32610, USA
Manuscript submitted July 31, 2018, accepted August 24, 2018
Short title: Cholecystitis as a Clue to Overlap Syndrome
doi: https://doi.org/10.14740/jmc3137w
| Abstract | ▴Top |
Lymphoplasmacytic cholecystitis (LPC) is a unique form of chronic cholecystitis, and it has been reported in patients with medical conditions such as primary sclerosing cholangitis, inflammatory bowel disease, and autoimmune pancreatitis. We report a case of LPC in a 23-year-old male with a history of autoimmune hepatitis who presented to the hospital with 3-day duration of abdominal pain, nausea and vomiting. A clinical diagnosis of acute cholecystitis and sclerosing cholangitis was made, and the patient was treated with laparoscopic cholecystectomy. Histopathological examination of the gallbladder revealed marked diffuse mononuclear inflammatory infiltrate that was rich in IgG4-positive plasma cells and focal neutrophilic inflammation with erosion. These findings were consistent with active LPC. The patient’s post-operative endoscopic retrograde cholangiopancreatography performed 1 month after cholecystectomy revealed typical features of primary sclerosing cholangitis. This is the first report of acute acalculous cholecystitis as an initial presentation of autoimmune hepatitis and primary sclerosing cholangitis overlap syndrome.
Keywords: Lymphoplasmacytic cholecystitis; Autoimmune hepatitis; Primary sclerosing cholangitis; Overlap syndrome; IgG4
| Introduction | ▴Top |
Lymphoplasmacytic cholecystitis (LPC) is an acalculous chronic inflammatory disease of gallbladder that was initially described in patients with primary sclerosing cholangitis [1-4]. Histologically, gallbladder with LPC has a triad of findings including diffuse chronic inflammatory infiltrate, a predominance of plasma cells in the lamina propria, and mucosal nodular lymphoid aggregate with occasional germinal center formation. An association of LPC with other forms of chronic pancreatobiliary disease, including neoplastic obstruction and autoimmune pancreatitis, has also been suggested [5-7]. In one recent study, LPC could occur as sporadic cases in older men and presented with typical signs and symptoms of acute cholecystitis such as fevers and/or leukocytosis [4]. LPC in patients with inflammatory bowel disease has been reported [8].
“Autoimmune cholecystitis” is probably an alternative term in the literature for some chronic cholecystitis morphologically resembling LPC. One study reported its association with autoimmune hepatitis and autoimmune pancreatitis [9]. In that case report, the patient experienced chronic vague abdominal pain, dark urine, acholic stools, and jaundice. Laboratory studies revealed elevated aspartate aminotransferase (AST), alanine aminotransferase (ALT), gamma-glutamyltransferase (GGT), direct bilirubin, positive anti-nuclear antibody (ANA), positive anti-smooth muscle antibody (ASMA), elevated IgG (4,080 mg/dL), and elevated lipase. Abdominal magnetic resonance imaging (MRI) revealed biliary duct dilatation with unremarkable pancreas. The patient had a liver biopsy showing chronic active hepatitis with mononuclear cell infiltrate and conspicuous plasma cells, features of autoimmune hepatitis. The portal inflammation relatively spared intrahepatic bile ducts. There was periductal inflammation but no obvious cholangitis, cholestasis or periductal fibrosis was seen. Histopathological examination of the gallbladder revealed features of LPC. Increased IgG4-positive plasma cells were noted in both liver biopsy and gallbladder; though the exact number of IgG4-positive plasma cells were not given in the report. The patient responded well with steroid and 6-mercaptopurine treatment after cholecystectomy, which led to normalization of his transaminases and lipase levels. In the report, autoimmune cholecystitis is thought to be the biliary manifestation of a multisystem fibroinflammatory disorder. However the association of LPC with autoimmune hepatitis alone has not been reported in the literature. Herein, in this case report, we describe one case of LPC developed 9 years after the initial diagnosis of autoimmune hepatitis and with a discussion of its clinical significance.
| Case Report | ▴Top |
The patient is a 23-year-old man with a past medical history significant for autoimmune hepatitis diagnosed 9 years ago that was based on clinical presentation, laboratory results and liver biopsy in an outside hospital. The patient was initially treated with corticosteroids with episodes of relapse when weaning the prednisone. The patient was later started on Imuran. Over the 9 years, he has had three liver biopsies with the most recent one done in October of 2016 revealing stage 1 fibrosis and mild steatosis, without significant ongoing autoimmune hepatitis. IgG4 level was within normal range. The patient is currently on Imuran (200 mg/day), Prograf (2 mg/day), prednisone (7.5 mg/day), and Ursodiol (600 mg/day). The patient is also an alpha-1 antitrypsin (A1AT) deficiency carrier, MZ phenotype. Since his last liver biopsy, the patient has had steadily increase in his alkaline phosphatase (ALP), transaminases AST and ALT, and total bilirubin (Table 1). He also has a 14-pound weight loss in the past 10 months.
 Click to view | Table 1. Laboratory Tests (Admitted in October 2017) |
The patient presented to our hospital for 3-day duration of abdominal pain, nausea and vomiting and was admitted. His abdominal pain got worse after he ate. He denied diarrhea, jaundice, itching, icterus, fever, chills, or sweats. Review of his systems was not remarkable. On physical examination, he had soft abdomen without distension, masses, or ascites. Tenderness to palpation in upper quadrants and epigastric area was present. He had normal bowel sounds. Laboratory studies on admission revealed normal electrolytes, blood glucose, and renal function tests. His liver function test revealed ALT at 78 IU/L, AST at 113 IU/L, ALP at 466 IU/L, total bilirubin at 10.2 mg/dL, direct bilirubin at 6.2 mg/dL, albumin at 4.2 g/dL, and total protein at 8.3 g/dL. ANA was positive at 1:80. IgG was slightly elevated at 1,647 mg/dL (reference: 635 – 1,616 mg/dL) (Table 1). Serum IgG4 was not tested on admission.
Several imaging studies on admission were performed. Right upper quadrant ultrasound revealed distended gallbladder with small sludge (Fig. 1a). The liver was echogenic and demonstrated beam attenuation consistent with fatty liver disease. There were no focal masses. The portal veins were non-dilated and demonstrated hepatopetal flow. The common bile duct was prominent and measured 6 mm in diameter. There’s no evidence of focal obstruction. The sonographic Murphy sign was equivocal due to pain medication. Computed tomography (CT) of the abdomen and pelvis with intravenous contrast revealed unremarkable liver without intrahepatic fracture pattern or biliary duct dilatation. The common bile duct appeared enhancing, suspicious for cholangitis (Fig. 1b). The gallbladder was markedly dilated with thickened wall. There was no definite pericholecystic fluid. A cholescintigraphy and hepatobiliary scintigraphy (HIDA) scan showed no filling of the gallbladder which was consistent with acute cholecystitis (Fig. 1c). Magnetic retrograde cholangiopancreatography (MRCP) plus pancreas without and with intravenous contrast revealed dilated gallbladder with mild associated stranding concerning for acute cholecystitis (Fig. 1c). No choledocholithiasis or dilation of common bile duct or pancreatic duct was noted on MRCP.
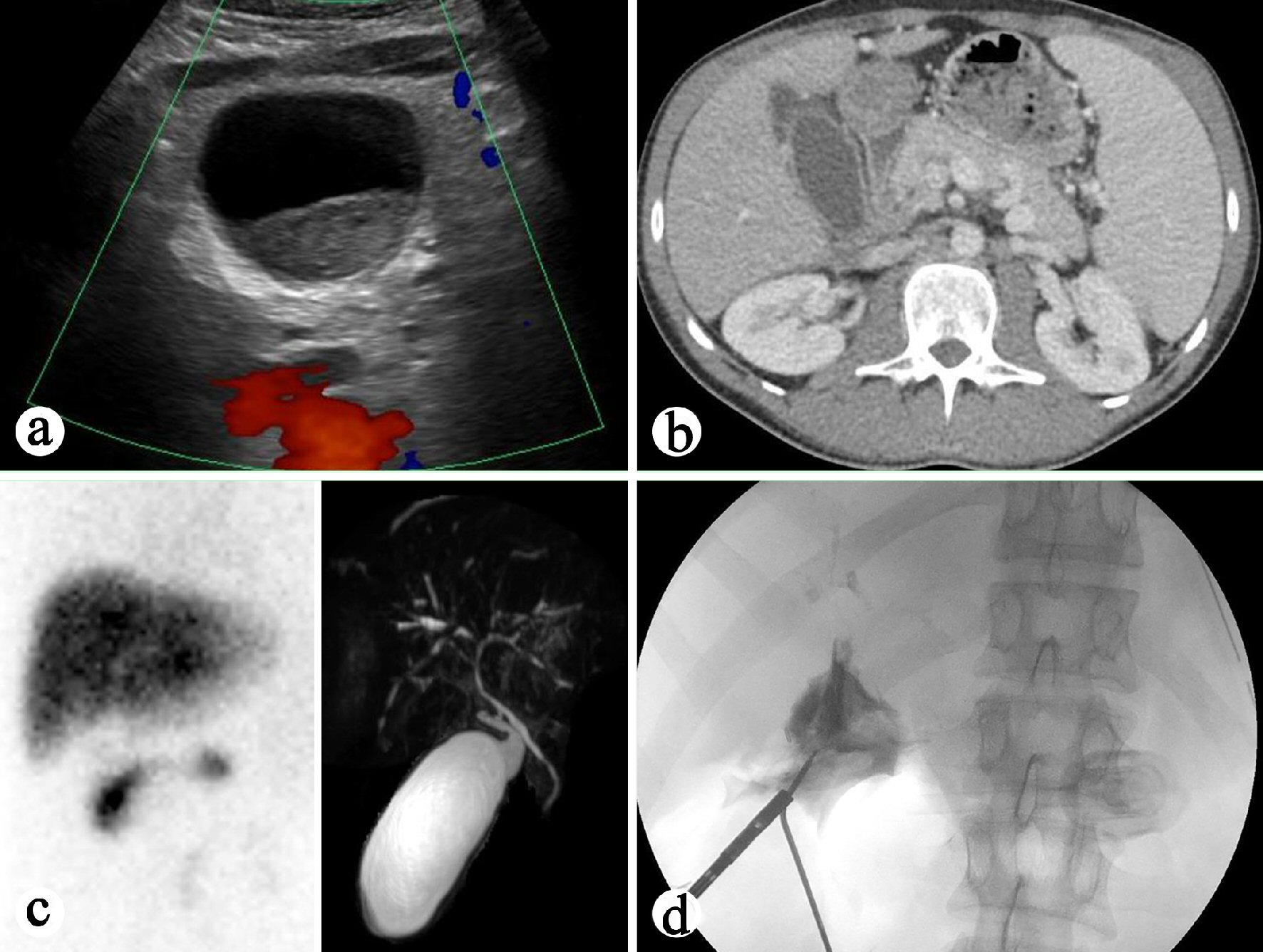 Click for large image | Figure 1. Preoperative and intraoperative imaging study of the gallbladder. (a) Right upper quadrant ultrasound showing distended gallbladder. (b) CT of the abdomen showing enhancing common bile duct. (c) Left: HIDA biliary study revealing no filling of the gallbladder. Right: MRCP demonstrating dilated gallbladder and mild associated stranding concerning for acute cholecystitis. (d) Intraoperative cholangiography revealing atretic common bile duct with moth-eaten appearance, features concerning for cholangitis. |
The patient was diagnosed with cholecystitis and sclerosing cholangitis and initially treated with Zofran and fluid bolus for 4 days. Laparoscopic cholecystectomy was then performed. Intraoperative cholangiography revealed that the common bile duct appeared atretic with “moth-eaten” appearance but without dominant stricture (Fig. 1d).
The cholecystectomy specimen consisted of an opened gallbladder with scattered areas of red-brown punctate hemorrhage on the outer surface. The cystic duct measured 0.7 cm in length and has a diameter of 0.6 cm. No calculi were identified in the gallbladder. The mucosal surface was pink-tan to pale-yellow with no distinct masses or lesions grossly identified. There was diffuse thickening of the gallbladder wall (0.4 - 0.9 cm). Histologically, all sections of the gallbladder revealed marked diffuse mononuclear inflammatory infiltrate that was plasma cell-rich (Fig. 2a, b). Focal neutrophilic inflammation with erosion was present. Focally prominent lymphoid aggregates were noted in the mucosa. Epithelial hyperplasia and multifocal intestinal metaplasia were present (Fig. 2c). The chronic cholecystitis involved the cyst duct resection margin. No granulomas, dysplasia, cholelithiasis, storiform fibrosis, or obliterative phlebitis were identified. Immunohistochemistry for IgG4 and IgG was performed, which revealed the presence of 25 - 60 IgG4-positive plasma cells per high power field but the ratio of IgG4 to IgG positive cells was 0.2 (Fig. 2d). A diagnosis of LPC was made based on the histology and an increased number of IgG4-positive plasma cells. In addition, to differentiate IgG4-related sclerosing cholangitis versus primary sclerosing cholangitis, recommendations to check serum IgG4 level and to perform endoscopic retrograde cholangiopancreatography (ERCP) were made to assess immune-mediated cholangitis in the remaining extrahepatic bile ducts.
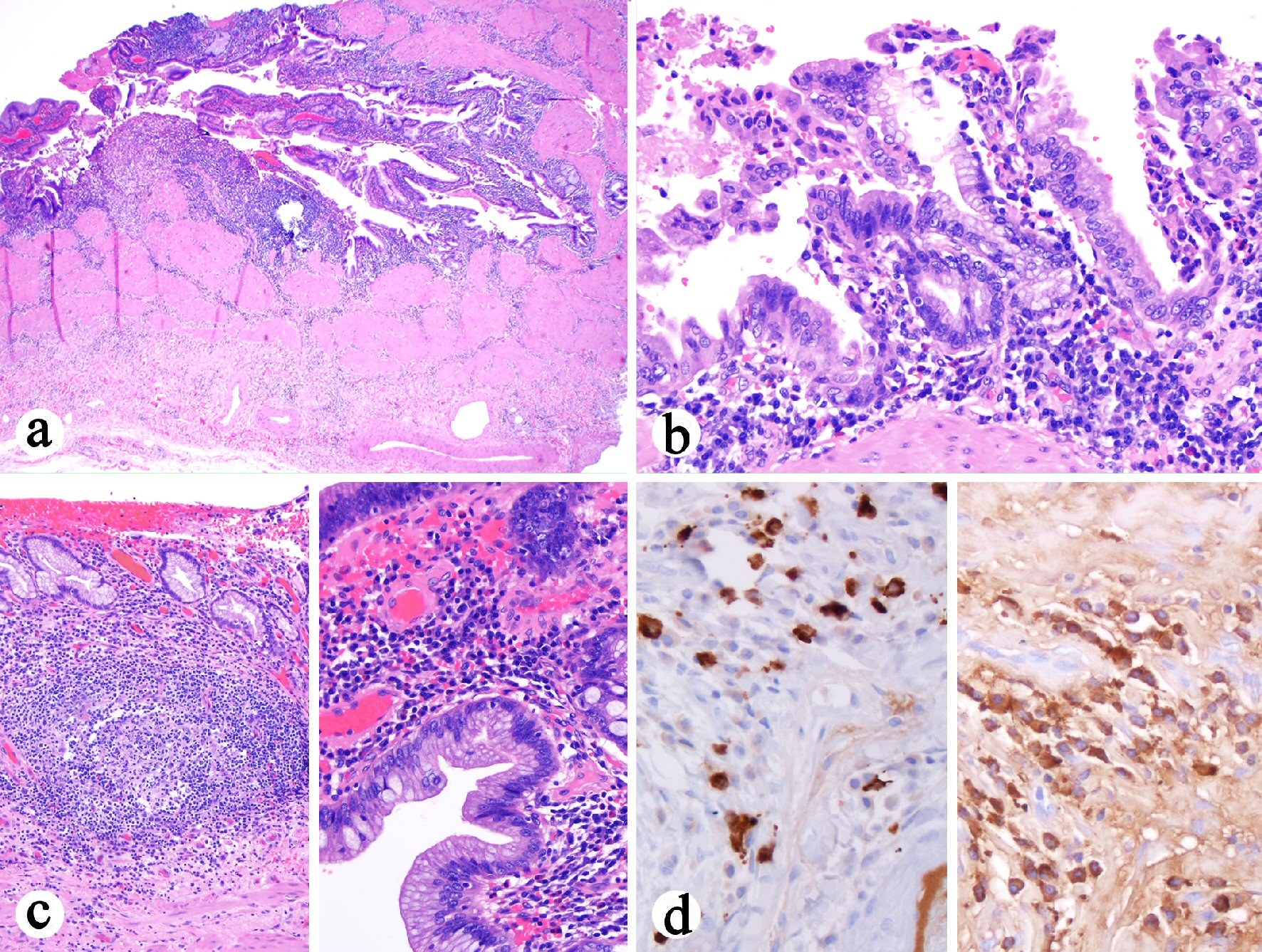 Click for large image | Figure 2. Histologic features of cholecystectomy. (a) Diffuse transmural mononuclear inflammatory infiltrate (H&E stain, 40 ×). (b) The plasma cell-rich mononuclear inflammatory infiltrate (H&E stain, 200 ×). (c) Left: Prominent lymphoid aggregates in the lamina propria (H&E stain, 100 ×). Right: The epithelium shows hyperplasia and focal intestinal metaplasia but negative for dysplasia (H&E stain, 400 ×). (d) Left: Increased number of IgG4 positive cells (immunostain, 400 ×). Right: IgG positive cell infiltrate in the wall (immunostain, 400 ×). The ratio of IgG4 to IgG positive cells in the photographed area was 0.2. |
The patient’s symptoms including abdominal pain, nausea and vomiting were improved postoperatively and was discharged on the second postoperative day. The patient’s post-operative ERCR revealed features consistent with primary sclerosing cholangitis including segmental strictures such as a hilar stricture and a dominant stricture at the left main duct, a chain of lakes appearance and diffuse attenuation of the intrahepatic bile ducts (Fig. 3). Biliary brushing cytology revealed ductal cells and reactive atypia without evidence of malignancy. At 3 months post-operative follow-up, the patient was doing well and his ALT, AST, total bilirubin, and ALP improved significantly although not completely normalized (Table 1).
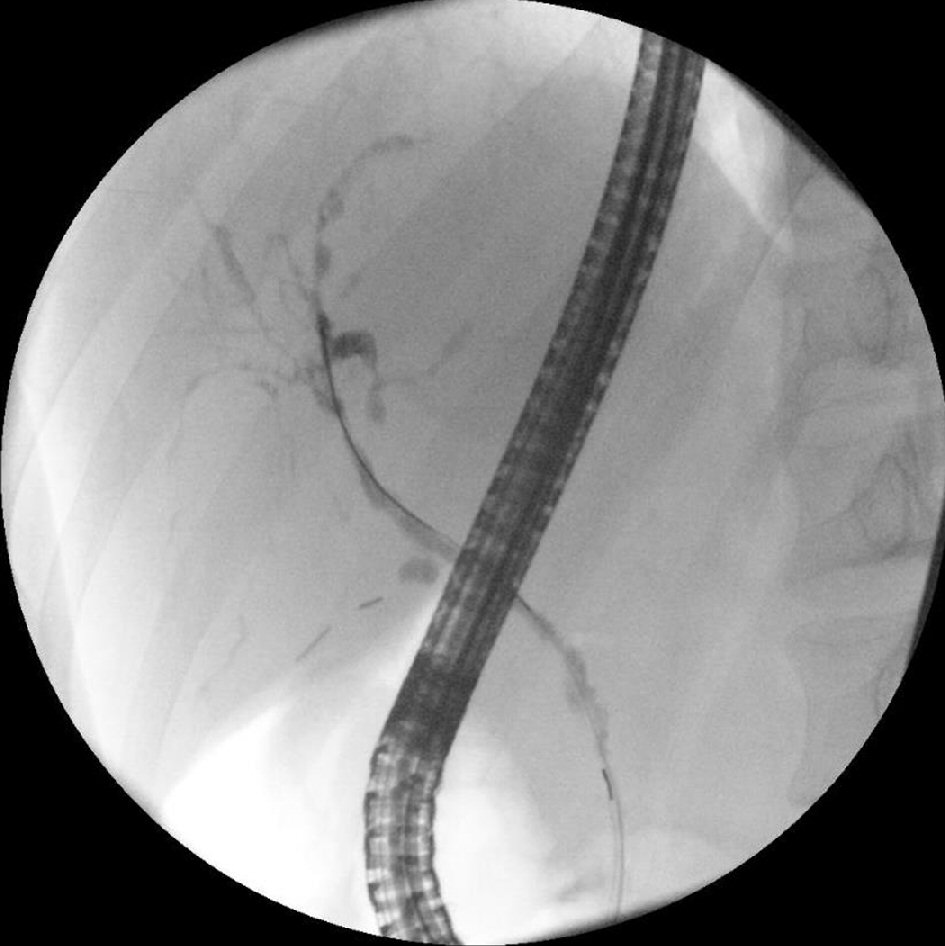 Click for large image | Figure 3. ERCR performed 1 month after cholecystectomy revealing a hilar stricture and a dominant stricture at the left main duct, a chain of lakes appearance and diffuse attenuation of the intrahepatic bile ducts, features of primary sclerosing cholangitis. |
| Discussion | ▴Top |
LPC is a unique form of chronic cholecystitis characterized by the presence of diffuse plasma cells-rich mononuclear inflammatory infiltrate and prominent lamina propria lymphoid aggregate. Gallstones are usually absent. LPC has been reported in patients with inflammatory bowel disease [8], primary sclerosing cholangitis [1-4], and autoimmune pancreatitis [5-7]. Recently LPC was reported as sporadic cases in old man who presented with fever and/or leukocytosis. A rare case of autoimmune cholecystitis with histology resembling LPC was reported in a patient with systemic autoimmune disease (autoimmune hepatitis/cholangitis and autoimmune pancreatitis) [9]. But LPC in patient with autoimmune hepatitis alone has not been reported.
Our case is unique. Clinically, the patient had symptoms of acute cholecystitis with 3-day duration of abdominal pain, nausea and vomiting. Eating aggravated the abdominal pain. The imaging studies including ultrasound, HIDA biliary scan, and abdominal CT revealed dilated gallbladder without evidence of choledocholiathiasis. The common bile duct was prominent on ultrasound and showed enhancement on CT imaging but no stricture noted; intrahepatic ducts were not dilated. His pancreas was normal. Intraoperative cholangiography revealed moth-eaten appearance for the common bile duct. Macroscopic examination of his gallbladder revealed diffuse wall thickening (0.4 to 0.9 cm). No gallstones or mass lesions were identified. No stricture was noted for the cystic duct. The serosa was smooth and glistening. All sections of the gallbladder showed typical features of LPC. But diagnostic features of classic IgG4-related autoimmune cholecystitis, i.e., storiform fibrosis and obliterating phlebitis, were not present. Immunohistochemistry revealed the presence of up to 60 IgG4 positive plasma cells per high power field, but the ratio of IgG4 to IgG-positive cells was 0.2. In addition, multifocal intestinal metaplasia was noted, which indicated a chronic process and was corresponding to the patient’s increased ALP and bilirubin in the past 10 months prior to the current acute presentation. The discrepancy between acute clinical presentation and chronic histological changes is similar to a recent report for this entity [4]. No dysplasia was identified in the gallbladder despite extensive sampling in our case. However the presence of gallbladder intestinal metaplasia has been associated with increased risk for gallbladder cancer [10].
In the current case, the chronic inflammatory process extended to the resection margin of the cystic duct. The fact that the patient had elevated ALP 1 year prior to the current admission raised a possibility that the current cholecystitis may be an extension or manifestation of primary sclerosing cholangitis. Retrospective review of previous liver biopsy from October 2016 (1 year prior to the presentation) revealed portal lymphocytic inflammation, bile duct injury, ductular proliferation, portal fibrosis, edema, lobular neutrophilic inflammation, and periportal hepatocyte resetting, subtle features of biliary obstruction (Fig. 4). The impression of cholangitis was further supported by radiological studies. ERCP performed 1 month after cholecystectomy confirmed the diagnosis of primary sclerosing cholangitis. In the context of previous history of autoimmune hepatitis, the patient’s currently liver disease fits best with autoimmune hepatitis and primary sclerosing cholangitis overlap syndrome. This is the first case report for acute acalculous cholecystitis as the initial presentation of autoimmune hepatitis and primary sclerosing cholangitis overlap syndrome.
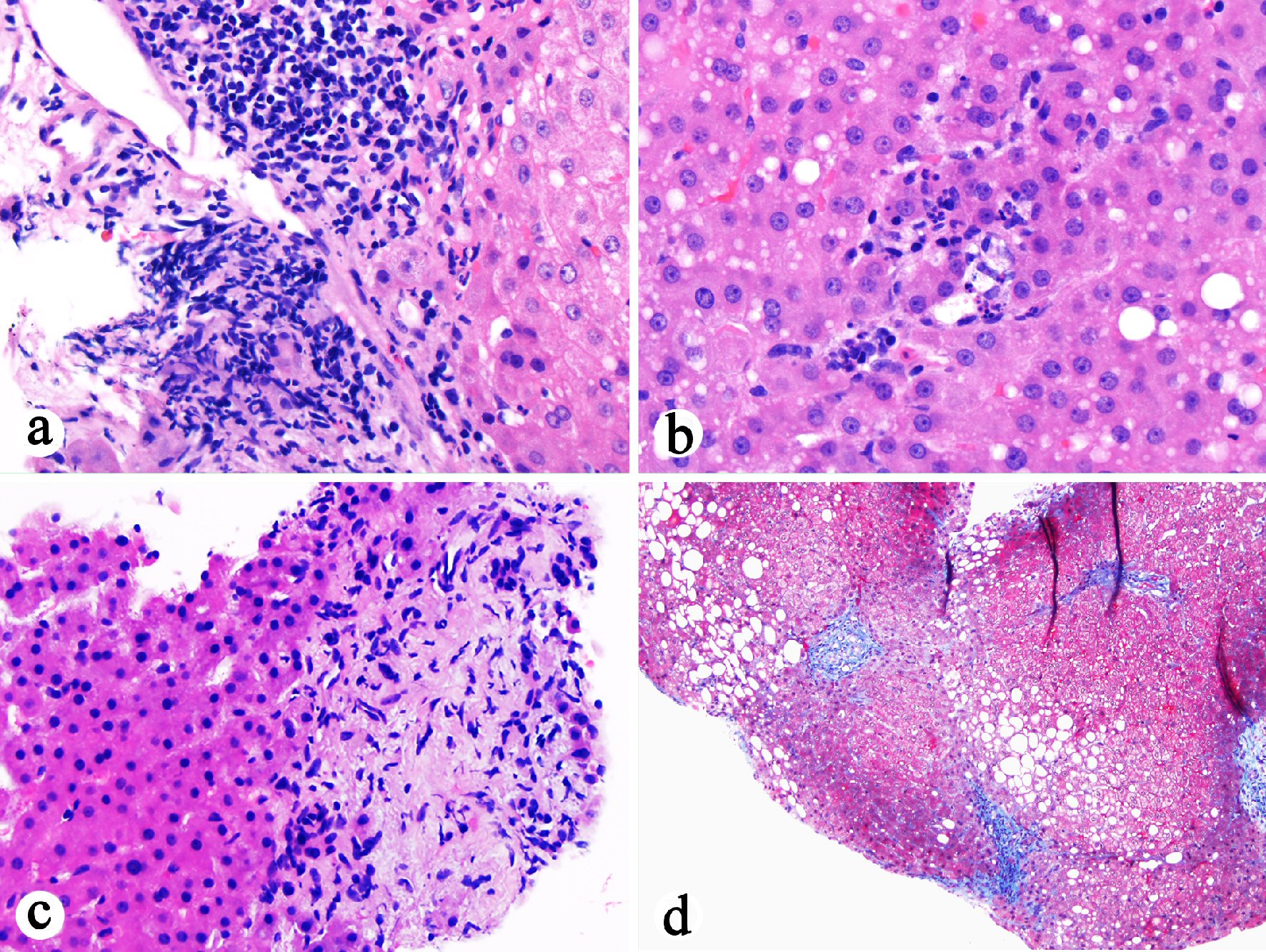 Click for large image | Figure 4. Histopathological examination of previous liver biopsy (October 2016) showing features suggestive of biliary flow impairment. (a) Portal lymphocytic inflammation, bile duct injury, portal fibrosis, and edema (H&E stain, 200 ×). (b) Lobular neutrophilic inflammation with associated hepatocyte apoptosis in addition to mild steatosis (H&E stain, 400 ×). (c) Portal ductular proliferation, portal fibrosis and edema, and periportal hepatocyte rosette formation (H&E stain, 400 ×). (d) Stellate portal fibrosis (trichrome stain, 100 ×). |
Our case report has several limitations. First, even though no gallstones were identified by imaging or by macroscopic examination of the gallbladder, this did not completely rule out cholelithiasis-associated cholecystitis as the gallstones may have passed out of the body through the intestines. Second, the presence of up to 60 IgG4-positive plasma cells per high power field in the gallbladder wall suggests an IgG4-related cholecystitis, but the ratio of IgG4 to IgG-positive cells were only 0.2 and serum IgG4 testing was not performed, thus this suspicion cannot be confirmed further. Third, the patient has not had colonoscopy yet and thus whether his LPC was related to idiopathic inflammatory bowel disease remained unclear at the time being.
In summary, we reported a case of LPC in a young adult patient developed 9 years after the initial diagnosis of autoimmune hepatitis. The diagnosis of LPC in this case prompted additional confirmatory study of the remaining bile duct; ERCP performed 1 month after cholecystectomy confirmed that the patient had primary sclerosing cholangitis. With all the available information, the patient’s current liver disease fits best with an overlap syndrome (autoimmune hepatitis with primary sclerosing cholangitis). This case report highlights the significance of LPC recognition by pathologist in young patients with autoimmune hepatitis. Large studies are needed to examine the prevalence of LPC in young patients with primary sclerosing cholangitis, as well as its predictive value.
Conflict of Interest
None.
Grant Support
None.
| References | ▴Top |
- Jessurun J, Bolio-Solis A, Manivel JC. Diffuse lymphoplasmacytic acalculous cholecystitis: a distinctive form of chronic cholecystitis associated with primary sclerosing cholangitis. Hum Pathol. 1998;29(5):512-517.
doi - Kamisawa T, Tu Y, Nakajima H, Egawa N, Tsuruta K, Okamoto A, Horiguchi S. Sclerosing cholecystitis associated with autoimmune pancreatitis. World J Gastroenterol. 2006;12(23):3736-3739.
doi pubmed - Said K, Glaumann H, Bergquist A. Gallbladder disease in patients with primary sclerosing cholangitis. J Hepatol. 2008;48(4):598-605.
doi pubmed - Hammer ST, Appelman HD. Sporadic lymphoplasmacytic cholecystitis: a clinicopathologic entity. Am J Clin Pathol. 2014;142(2):209-212.
doi pubmed - Abraham SC, Cruz-Correa M, Argani P, Furth EE, Hruban RH, Boitnott JK. Diffuse lymphoplasmacytic chronic cholecystitis is highly specific for extrahepatic biliary tract disease but does not distinguish between primary and secondary sclerosing cholangiopathy. Am J Surg Pathol. 2003;27(10):1313-1320.
doi pubmed - Abraham SC, Cruz-Correa M, Argani P, Furth EE, Hruban RH, Boitnott JK. Lymphoplasmacytic chronic cholecystitis and biliary tract disease in patients with lymphoplasmacytic sclerosing pancreatitis. Am J Surg Pathol. 2003;27(4):441-451.
doi pubmed - Wang WL, Farris AB, Lauwers GY, Deshpande V. Autoimmune pancreatitis-related cholecystitis: a morphologically and immunologically distinctive form of lymphoplasmacytic sclerosing cholecystitis. Histopathology. 2009;54(7):829-836.
doi pubmed - Lin J, Shen B, Lee HJ, Goldblum JR. Histopathological characterization of cholecystectomy specimens in patients with inflammatory bowel disease. J Crohns Colitis. 2012;6(9):895-899.
doi pubmed - Memon Z, Moya DA, Baker R, Kozielski R, Baker S. Autoimmune cholecystitis. J Pediatr Gastroenterol Nutr. 2012;54(4):441.
doi pubmed - Lewis JT, Talwalkar JA, Rosen CB, Smyrk TC, Abraham SC. Prevalence and risk factors for gallbladder neoplasia in patients with primary sclerosing cholangitis: evidence for a metaplasia-dysplasia-carcinoma sequence. Am J Surg Pathol. 2007;31(6):907-913.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.