

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 9, Number 12, December 2018, pages 406-408
Pancreatic Neuroendocrine Tumor Invading the Stomach and Spleen: A Case Report
Eric Gladstonea, c, Juskaran Chadhab
aDepartment of Medicine, Lenox Hill Hospital, New York, NY, USA
bDepartment of Hematology and Oncology, Lenox Hill Hospital, New York, NY, USA
cCorresponding Author: Eric Gladstone, Department of Medicine, Lenox Hill Hospital, New York, NY, USA
Manuscript submitted November 13, 2018, accepted November 21, 2018
Short title: PNET Invading the Stomach and Spleen
doi: https://doi.org/10.14740/jmc3216
| Abstract | ▴Top |
Pancreatic neuroendocrine tumors are a rare group of heterogeneous neoplasms. Approximately half have metastasized by the time of diagnosis, most commonly to the liver. We present the case of an 88-year-old female who presented with syncope. Imaging revealed a large pancreatic mass. She was found to have a pancreatic neuroendocrine tumor directly invading the stomach and spleen with liver and lymph node metastases. This case demonstrates the rare occurrence of gastric invasion by a pancreatic neuroendocrine tumor.
Keywords: Pancreatic neuroendocrine tumor; Invasion; Spleen; Stomach; Multivisceral resection
| Introduction | ▴Top |
Pancreatic neuroendocrine tumors (PNETs) arise from the hormone-secreting islet cells, and account for 1-2% of all pancreatic neoplasms. The most common site of PNET metastases is to the lymph nodes at 50%, followed by liver at 30% [1]. Liver metastasis is the most common cause of death in PNET. Metastasis to the spleen is observed in 10% [1]. Gastric involvement is less common and the precise incidence is not reported. We present the case of a PNET invading both the stomach and spleen.
| Case Report | ▴Top |
An 88-year-old female with a past medical history of diet-controlled type 2 diabetes mellitus, hypothyroidism, hyperlipidemia, osteoporosis and unknown cardiac arrhythmia presented to the emergency department (ED) for witnessed syncopal episode. She was found to have orthostatic hypotension in the setting of poor PO intake. She reported 2 years of intermittent, non-localized abdominal discomfort without other associated gastrointestinal (GI) symptoms. Fifteen months prior she had a CT abdomen at an outside facility which she reported showed a small abdominal “lesion” that was never fully investigated.
CT abdomen in the ED showed a 5.9 × 6.5 × 5.1 cm mass near the splenic hilum involving the pancreatic tail extending to the spleen. The mass occluded the splenic artery and vein resulting in splenic infarcts. There was also a 4 × 3.3 × 2.8 cm para-aortic heterogeneous mass and several low-density hepatic lesions measuring up to 3.5 cm (Fig. 1).
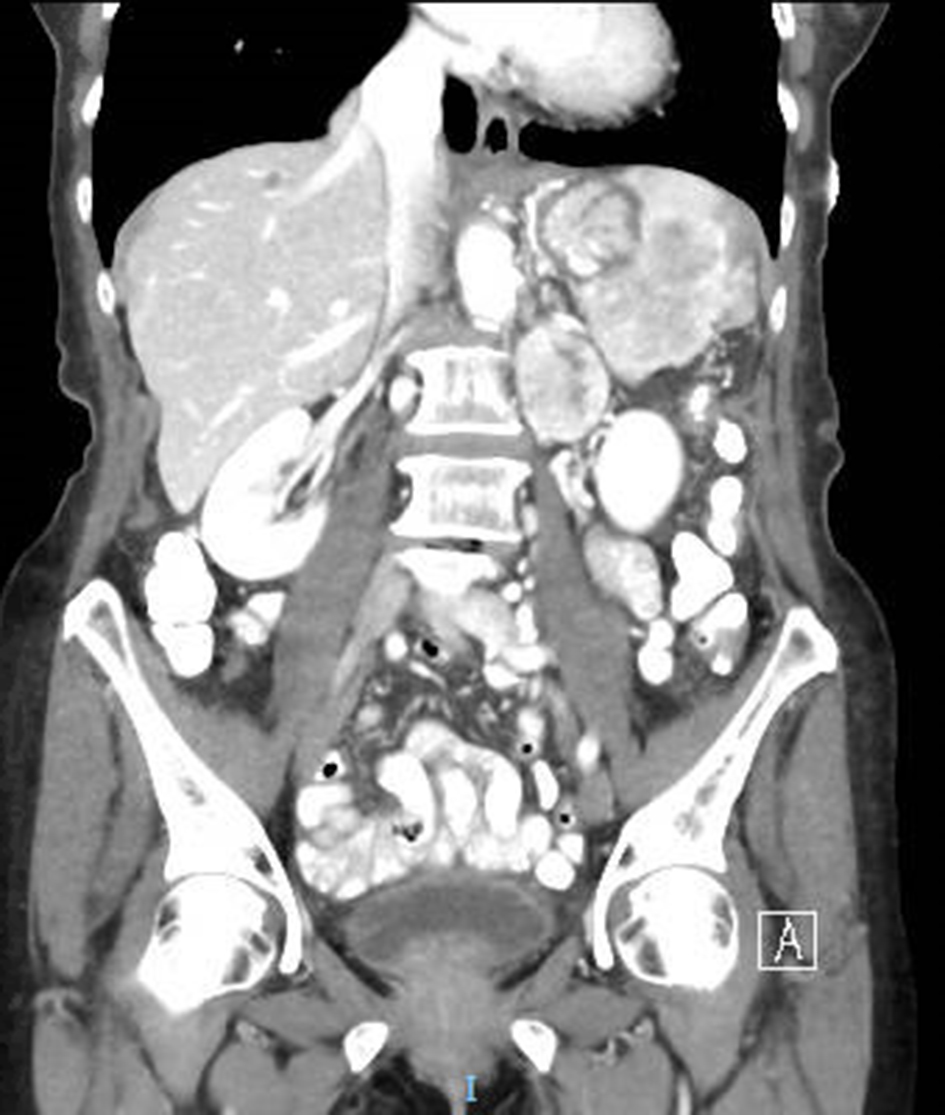 Click for large image | Figure 1. Coronal section on CT abdomen. CT revealed multiple splenic infarcts, a mass in the pancreatic tail and splenic hilum, as well as a para-aortic mass. |
Serum chromogranin A (CgA) level was elevated at 447 ng/mL. Carbohydrate antigen (CA) 19-9, carcinoembryonic antigen (CEA) and lactate dyhydrogenase (LDH) were within normal limits. Core biopsy of the para-aortic mass revealed well-differentiated neuroendocrine tumor (NET). The tumor cells were positive for synaptophysin, chromogranin and pancytokeratin. The Ki-67 was less than 3%. Somatostatin receptor positron-emission tomography (PET) imaging showed intense radiotracer uptake in the previously-observed pancreatic, para-aortic and hepatic masses (Fig. 2).
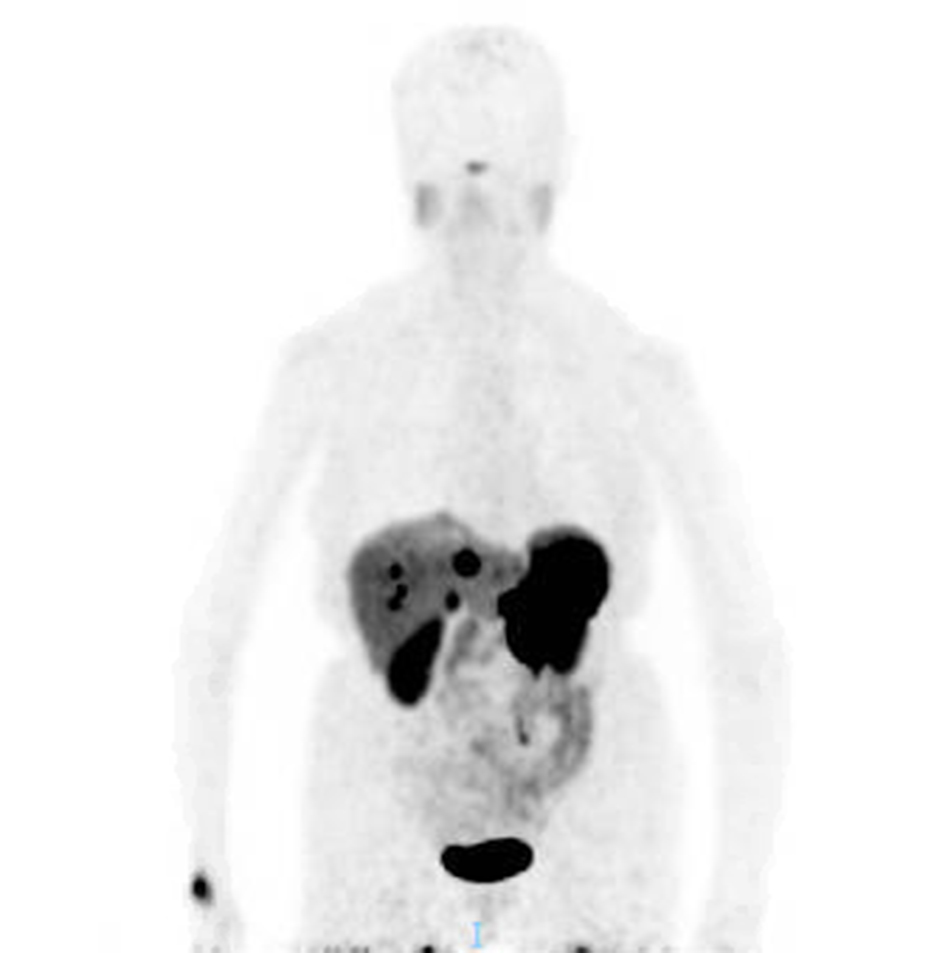 Click for large image | Figure 2. Somatostatin receptor PET imaging. Increased radiotracer uptake was observed in the pancreatic, hepatic and para-aortic masses. |
The patient underwent distal subtotal pancreatectomy, splenectomy, partial gastrectomy, cholecystectomy and resection of both of two liver lesions. The liver lesions and para-aortic mass were metastatic NETs. There was extensive venous invasion of medium-sized veins at the pancreatic margin. The tumor infiltrated the spleen (involving 60% of its pulp) and stomach (involving 10% of its serosa). One of 13 examined gastric regional lymph nodes was involved. The tumor exhibited average mitotic rate of 9 mitoses/mm2, and proliferative index of 15% by Ki67. Pathology revealed a grade 2 tumor with pathological stage classification pT4, pN1 and pM1a. The patient tolerated the procedure well and was discharged on post-operative day 6. She was followed up at an outside facility.
| Discussion | ▴Top |
The pancreas is an organ with both exocrine and endocrine functions. The cells of the exocrine pancreas produce digestive enzymes and tumors arising from these cells are generally adenocarcinomas. The endocrine functions are served by the hormone-secreting islet cells. These cells compose 1-2% of the pancreas by volume. PNETs arise from the hormone-secreting islet cells, and account for 1-2% of all pancreatic neoplasms. These tumors may also be referred to as pancreatic islet cell tumors. The incidence of PNET, estimated at < 1/100,000 in the USA, has slowly risen over recent decades due in part to increased awareness and improved imaging modalities [2]. PNET can be functioning (hormone-hypersecreting) or non-functioning. Insulinomas are the most common functioning PNETs, followed by gastrinomas, glucagonomas, somatostatinomas and VIPomas [3]. Patients with functioning PNET commonly present in earlier stages with symptoms due to hormone hypersecretion. Conversely, patients with non-functioning PNET typically present with abdominal pain or nausea when the tumor is larger. Patients with functional PNET typically have a favorable prognosis because they are diagnosed at earlier stages. Approximately 90% of PNETs are sporadic, whereas 10% are observed in patients with a familial endocrine tumor syndrome [3]. These syndromes include multiple endocrine neoplasia 1 (MEN1), tuberous sclerosis, neurofibromatosis type 1 and von Hippel-Lindau disease. The average time between tumor development and diagnosis has been reported at 5 - 10 years. Compared to exocrine tumors, PNETs have a favorable prognosis. Nonetheless, excluding insulinomas, 50-60% of PNETs have metastasized by the time they are diagnosed [4].
Splenic metastasis from any primary tumor is a rare phenomenon. It is hypothesized that the splenic microenvironment is hostile to tumor cells. Splenic metastases are usually indicative of widespread disease. Both metastasis and invasion of PNET to the spleen have been reported [5, 6]. Invasion of the splenic vein appears to be more common than to the pulp. Invasion of the splenic vein may account for one route of hematogenous spread. In one series of 273 patients with PNETs, 17% had preoperative CT evidence of major vascular involvement [7]. The portal vein was most commonly involved; 1.4% involved the splenic vein. Occlusion of the splenic vein predisposes to splenic infarction. Infarction can be caused by extrinsic compression of the vessel, torsion of the vessel or tumor invasion into the vessel. Infarction has led to splenic rupture in several reported cases of pancreatic adenocarcinoma [8]. In the case of our patient, CT demonstrated multiple splenic infarcts.
Numerous circulating biomarkers have been studied in PNET. CgA, 5-hydroxyindoleacetic acid (5-HIAA) and neuron-specific enolase (NSE) have perhaps been studied most extensively. Measurement of CgA level is clinically useful. CgA is a secretory protein produced by NET cells. Unlike 5-HIAA, CgA is released by both functioning and nonfunctioning tumors. Elevations in NSE typically reflect high tumor burden, somewhat limiting its utility. The degree of CgA level elevation is correlated to extent of disease. Furthermore, trending CgA is useful in both monitoring recurrence and response to therapy. Of note, renal failure and proton pump inhibitors are associated with artificially elevated levels of CgA. In this case, somatostatin receptor PET imaging contributed to the diagnosis of NET.
The Ki-67 index and mitotic count are used to classify and prognosticate PNET. Tumor nomenclature follows the 2010 WHO classification which distinguishes NET G1, NET G2, neuroendocrine carcinoma and mixed adenoneuroendocrine carcinoma. Grade 1 is defined as mitotic count < 2 per 10 high-power fields (HPF) or Ki-67 index < 3%. Grade 2 is defined as mitotic count between 2 and 20 per 10 HPF or Ki-67 index between 3% and 20%. Neuroendocrine carcinoma is defined as mitotic count > 20 per 10 HPF or Ki-67 index > 20%. It is important to recognize that significant discordance can exist in the Ki-67 index between samples taken from the same tumor. This can be multifactorial, but in many instances, is due to intratumoral heterogeneity. When discrepancy exists between grading based on mitotic count and Ki-67 index, the higher grade is used.
The treatment strategy of PNET generally requires multidisciplinary care. The expertise of medical, radiation and surgical oncology, as well as endocrinology, gastroenterology and interventional radiology, may each be of use. Surgery with curative intent is first-line treatment in low-to-intermediate grade tumors. Surgical resection of metastases is performed when possible. There are numerous options for medical management. First-line treatment are the somatostatin analogs which include octreotide and lanreotide. Adjuvant agents include everolimus, sunitinib, interferon (IFN)-α, bevacizumab and cytotoxic agents (in advanced disease). Platinum-based chemotherapy is first-line treatment for metastatic high-grade tumors.
In this case, the patient was treated surgically. It is unknown what medical treatments if any she may have received following discharge. Given her metastases (including lymph node), discussion should have been held about adjuvant therapy. Once she was transferred to the surgical service, it became unclear from chart review what medical treatments she was offered. It would have behooved the treatment team to have presented the case at a tumor board.
Her case represents a common presentation for a PNET, with some exceptions. The fact that she presented with dehydration is not unusual for PNET. This was likely partly due to her diminished appetite and progressive vague abdominal discomfort. She experienced no further syncopal episodes following her initial presentation. It should also not be surprising that her disease was metastatic at the time of discovery. It would be informative to know exactly what was observed on her abdominal CT scan 15 months prior. It is unknown over what period of time her disease progressed. The sequence of progression is also unknown in this case. We speculate that gastric invasion was a later event. Anecdotally, gastric invasion appears uncommon when compared to hepatic and even splenic involvement.
Whether her tumor was functional or non-functional also remains unknown. She did not demonstrate obvious symptoms of a functional PNET; however, testing for hormone hypersecretion was not performed. More relevant to her prognosis is the fact that the tumor grade was upstaged from G1 to G2 based on surgical pathology. While the Ki-67 index was < 3% on initial core biopsy of a para-aortic lymph node metastasis, the Ki-67 index of the primary tumor was 15%. As mentioned previously, discrepancy in staging based on Ki-67 index is not an unusual phenomenon. Consensus does not yet exist on exactly which factors are best for prognostication. Our understanding of PNET behavior is rapidly evolving. As PNET awareness and detection continue to increase, additional insight will be gained.
Conflict of Interest
We herein declare that there is no financial support or relationships that may pose conflict of interest.
Grant Support
None.
Author Contributions
EG was involved in patient care and writing the manuscript. JC was involved in writing and revision of the manuscript. Each author has reviewed the final version of the manuscript and approves it for publication.
Patient Consent
Consent was obtained from the patient.
| References | ▴Top |
- Herlin C, Yachouh J, Journet JL, Goudot P. Extramandibular position of the inferior alveolar nerve. Analysis and review of the literature. Surg Radiol Anat. 2013;35(9):811-815.
doi pubmed - McKenna LR, Edil BH. Update on pancreatic neuroendocrine tumors. Gland Surg. 2014;3(4):258-275.
pubmed - Pea A, Hruban RH, Wood LD. Genetics of pancreatic neuroendocrine tumors: implications for the clinic. Expert Rev Gastroenterol Hepatol. 2015;9(11):1407-1419.
doi pubmed - Ellison TA, Edil BH. The current management of pancreatic neuroendocrine tumors. Adv Surg. 2012;46:283-296.
doi - Shah SA, Amarapurkar AD, Prabhu SR, Kumar V, Gangurde GK, Joshi RM. Splenic mass and isolated gastric varices: a rare presentation of a neuroendocrine tumor of the pancreas. JOP. 2010;11(5):444-445.
pubmed - Sakuma Y, Yasuda Y, Sata N, Hosoya Y, Shimizu A, Fujii H, Matsubara D, et al. Pancreatic neuroendocrine tumor with metastasis to the spleen: a case report. BMC Cancer. 2017;17(1):37.
doi pubmed - Norton JA, Harris EJ, Chen Y, Visser BC, Poultsides GA, Kunz PC, Fisher GA, et al. Pancreatic endocrine tumors with major vascular abutment, involvement, or encasement and indication for resection. Arch Surg. 2011;146(6):724-732.
doi pubmed - Smith WM, Lucas JG, Frankel WL. Splenic rupture: a rare presentation of pancreatic carcinoma. Arch Pathol Lab Med. 2004;128(10):1146-1150.
pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.