

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 10, Number 7, July 2019, pages 209-217
Report of Seven Cases of Langerhans Cell Histiocytosis in a Single Hospital in Japan
Takuji Tanakaa, b, Riyoko Niwaa, Fumimasa Etoria, Asuka Ohashia, Ryogo Aokia, Naomi Kawaguchia, Toshimasa Sakakimaa, Rina Miyamaea, Masashi Matsuyamaa, Naoki Watanabea
aDepartment of Diagnostic Pathology (DDP) & Research Center of Diagnostic Pathology (RC-DiP), Gifu Municipal Hospital, 7-1 Kashima-cho, Gifu 500-8513, Japan
bCorresponding Author: Takuji Tanaka, Department of Diagnostic Pathology (DDP) & Research Center of Diagnostic Pathology (RC-DiP), Gifu Municipal Hospital, 7-1 Kashima-cho, Gifu City, Gifu 500-8513, Japan
Manuscript submitted June 4, 2019, accepted July 19, 2019
Short title: Diagnosis of LCH in a Single Hospital
doi: https://doi.org/10.14740/jmc3325
| Abstract | ▴Top |
Langerhans cell histiocytosis (LCH), which shows a variable clinical presentation, is a disease that involves the proliferation of histiocytes. LCH occurs at any age and can affect a single system or multiple systems. The successful and accurate diagnosis of cases involving multiple organs requires a multidisciplinary approach. Since LCH receives limited attention, failure to recognize this disease in the early pediatric age can result in progression into adulthood. This study presents the clinical manifestations, imaging findings, pathological and cytological features, treatment strategies and prognoses of seven cases of LCH that were managed in a single hospital over the last 12 years. The ages of the patients ranged from 1 to 58 years. The sites at which LCH developed included the skin, lung, bone, lymph nodes and brain: one patient (adult male) had single-system/single-site LCH, three patients (adult male, adult female and girl) had single-system/multi-site LCH and three patients (adult female and two girls) had multi-system/multi-site LCH. BRAFV600E mutation was suggested in an adult female with multi-system/multi-site LCH and an adult male with single-system/multi-site LCH. We also discuss the pathogenesis and review the relevant literature.
Keywords: Langerhans cell histiocytosis; Histopathology; Cytopathology; Immunohistochemistry; Immunocytochemistry; Bone; Lung; Skin; Lymph node; Brain
| Introduction | ▴Top |
Langerhans cells (LCs) are dendritic cells that are located within the epidermis, mucosa, or bronchial epithelium, expressing CD1a and containing Birbeck’s granules [1]. Local inflammation is able to activate LCs and promote the migration of LCs to draining lymph nodes, resulting in differentiation into interdigitating cells. The first description of Langerhans cell histiocytosis (LCH) goes back to the late 19th century [2]. The clinical presentation of LCH is variable and LCH develops at any age and at any site (single-system or multi-system and single-site or multi-site (Table 1)); however, it more frequently occurs in the pediatric population [3-6]. The etiology of LCH remains unclear and has long been suspected to involve a combination of factors, including immune dysregulation, inflammation and malignancy. Thus, its rarity and the extreme heterogeneity of its clinical spectrum and natural course may disturb progress in understanding LCH.
 Click to view | Table 1. Clinical Classifications of LCH |
The discovery of the mutations in certain genes, including BRAFV600E and MAP2K1 that can be detected in approximately 75% of LCH patients, has suggested that the disease has a neoplastic nature [3, 7-11]. Children with LCH usually show a more aggressive clinical course that requires systemic chemotherapy [12]. Patients with high-risk LCH (Table 2) have a survival rate of almost 80% [9]. On the other hand, low-risk LCH patients (Table 2) have an excellent prognosis, with a long-term survival rate as high as 99% [9]. Adult patients usually present with limited skin or bone involvement [9]. They can be treated with surgical resection or focal radiation therapy, which result in an overall survival rate of 100% [4]. Smoking cessation can result in the spontaneous remission of pulmonary LCH [3]. Targeted therapy using certain BRAF inhibitors has been applied in selected patients with LCH [7].
Click to view | Table 2. Stratification of MS-LCH |
Our understanding of LCH has improved in the last 20 years [4, 6-8, 10, 13, 14]. Available treatment regimens can control the disease in the majority of patients [4, 6-8, 10, 13, 14]. The discovery of novel driver mutations and the development of targeted therapy promise better outcomes with fewer long-term therapy-related adverse events, especially for pediatric and adolescent patients [7, 9].
LCH can be diagnosed based on histopathological and/or cytological findings: the lesion usually consists of histiocytes, including characteristic LCs and multinucleated giant cells, with varying proportions of inflammatory cells, including eosinophils, macrophages and T-cell lymphocytes. The presence of pathognomonic Birbeck’s granules in LCH cells on electron microscopy and the detection of S100, langerin (CD207) and CD1a on their membrane are diagnostic. Because of the diversity of symptoms, the diagnosis of LCH is often made after a considerable delay. To better understand this rare disease, and to provide information that may facilitate an early diagnosis and appropriate treatment, we report seven cases of LCH that were encountered in our hospital over the last 12 years and present the diagnostic imaging, cytology and histopathology findings. The patients include four adults (two females and two males) and three infants (girls).
| Case Reports | ▴Top |
The present retrospective study was approved by the Medical Ethics Committee of the Gifu Municipal Hospital and was conducted in compliance with the tenets of The Declaration of Helsinki.
Patients (n = 7) who were diagnosed LCH and treated over the last 12 years (from October 25, 2005 to February 25, 2019) at Gifu Municipal Hospital were enrolled in the current study. A comprehensive retrospective review was conducted to evaluate the clinical, imaging, cytological and pathological characteristics of LCH. Information regarding the clinical manifestations and treatment was collected from the patients’ medical records. The computed tomography (CT) and/or magnetic resonance imaging (MRI) findings were acquired from the database at Gifu Municipal Hospital. Hematoxylin and eosin (H&E)-, Papanicolaou-, Giemsa- and periodic acid-Schiff (PAS)-stained slides and immunohistochemical and/or immunocytochemical slides were available for all but one of the cases. The diagnosis of LCH was confirmed based on a re-review of these slides by experienced pathologists (TT and NW) at Gifu Municipal Hospital. Furthermore, the PubMed database was searched for relevant studies published in the English language from 1963 to the present (February 25, 2019). After the exclusion of articles that were too generalized, the relevant literature was reviewed.
Seven cases of LCH we experienced over the last 12 years are listed in Table 3. We diagnosed lung LCH by bronchoalveolar lavage fluid (BALF) cytology alone in a single case (case 2). The patient was a young female smoker of 28 years of age.
Click to view | Table 3. Characteristics of Seven Cases of LCH |
Case 1
In June 2016, a 35-year-old female patient with a Brinkman index of 150 (10 cigarettes/day for 15 years) complained of severe back pain and the presence of a number of small cavitary and nodular lesions in the bilateral upper lobes was suggested by CT (Fig. 1a). Her back pain was considered to be caused by an osteolytic lesion of the left IV rib. BALF cytology and transbronchial biopsy (TBB) examinations revealed LCH in her lungs. In the BALF from the upper lobes (Fig. 1b, c), a number of histiocytes (64.3%) that were positive for CD1a (Fig. 1d) and S100 (Fig. 1e) antibodies were present in conjunction with neutrophils (3.5%), eosinophils (9.8%) and lymphocytes (22.4%). Granulomatous lesions containing histiocytes and eosinophils were present in the TBB specimens (Fig. 1f). These histiocytes with a slightly eosinophilic cytoplasm had the appearance of typical LCs with a distinct folded or lobulated, and often kidney-shaped nucleus. Nucleoli were not prominent. They were immunohistochemically positive for CD1a (Fig. 1g) and S100 (Fig. 1h). Their cytoplasm was also immunohistochemically positive for BRAFV600E antibody (Fig. 1i). Based on the cytological and histopathological findings in conjunction with those of immunocytochemical and immunohistochemical analyses, LCH of the lung and rib was diagnosed. The LCH lesions in both tissues disappeared following 6 months of smoking cessation and prednisolone treatment.
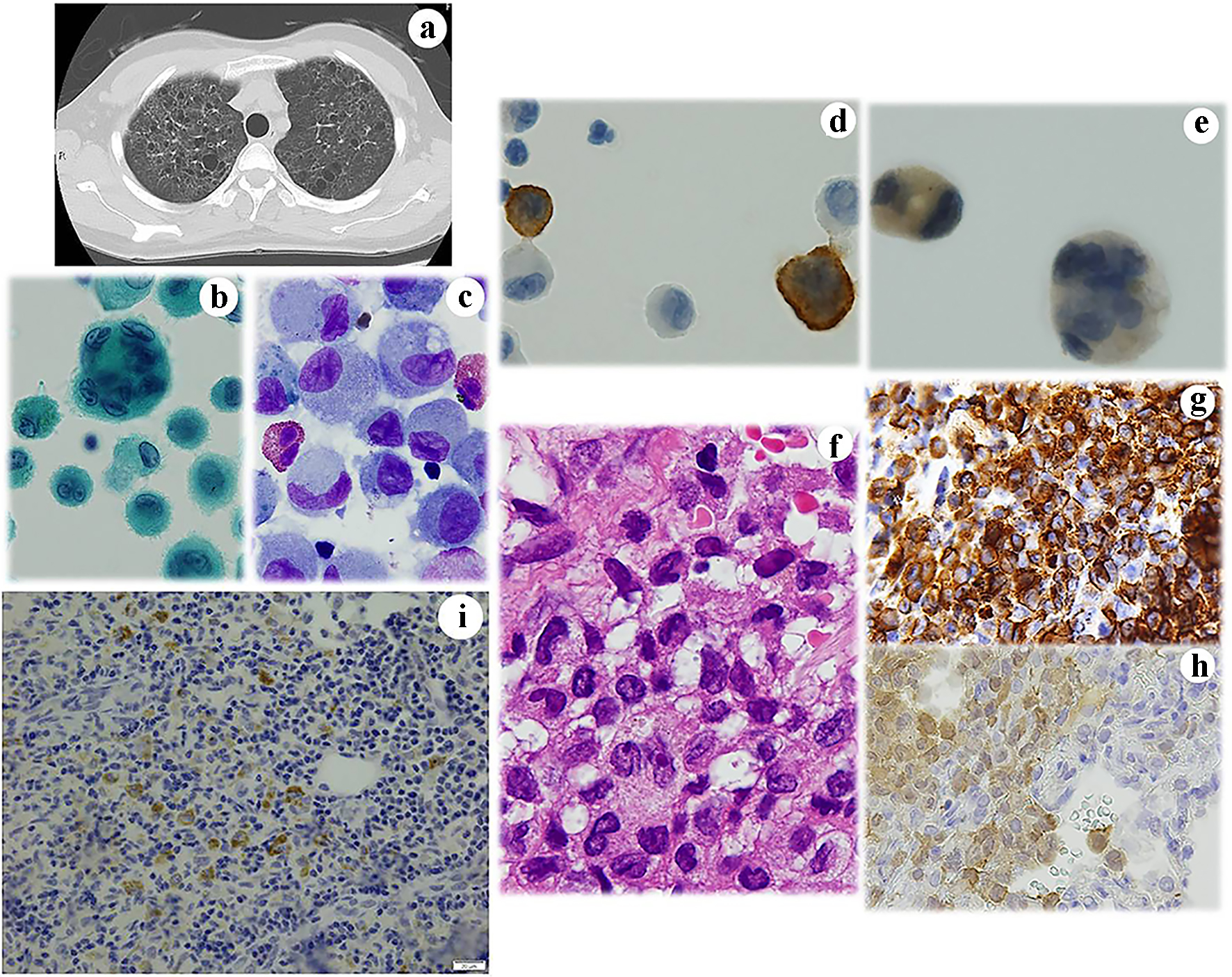 Click for large image | Figure 1. Case 1. (a) Chest CT shows small cystic lesions in the bilateral lungs. BALF cytology. Note the presence of large histiocyte-like cells with (b) multiple nuclei and (c) prominent grooved nuclei. These cells are immunocytochemically positive for (d) CD1a and (e) S100. Histopathologically, the lesion contains (f) abnormal histiocytes with grooved nuclei. Immunohistochemistry reveals that they are strongly positive for (g) CD1a and weakly positive for (h) S100. Some are positive for BRAFV600E antibodies. (b) Papanicolaou staining, × 400; (c) Giemsa staining, × 400; (d) CD1a immunocytochemistry; (e) S100 immunocytochemistry, × 400; (f) Hematoxylin and eosin staining, × 400; (g) CD1a immunohistochemistry, × 400; (h) S100 immunohistochemistry; and (i) BRAFV600E immunohistochemistry. CT: computed tomography; BALF: bronchoalveolar lavage fluid. |
Case 2
The patient was a 28-year-old woman with a Brinkman index of 165 (15 cigarettes/day for 11 years) who had a number of cavitary lesions in both lungs. The lesions were detected in September 2017 by CT during a health examination (Fig. 2a). A cytological examination of BALF revealed numerous atypical histiocytes with large cytoplasm and mono-nuclei or multi-nuclei (Fig. 2b, c). The nuclei were indented and convoluted with a coffee bean-like groove. Immunohistochemistry revealed that the cytoplasm was positive for S100 (Fig. 2d). Bronchofiberscopy showed increased CD1a-positive cells in the BALF. In the background, inflammatory cells, including eosinophils, were noted. In the BALF, 18.1% of the cells were CD1a-positive (370 cells) and CD4/CD8 ratio was 0.65. These findings strongly suggested lung LCH. At 8 months after the cessation of smoking, the cavity lesions had disappeared, the number of CD1a-positive cells decreased, and the CD4/CD8 ratio improved. Thus, we diagnosed the patient with lung LCH.
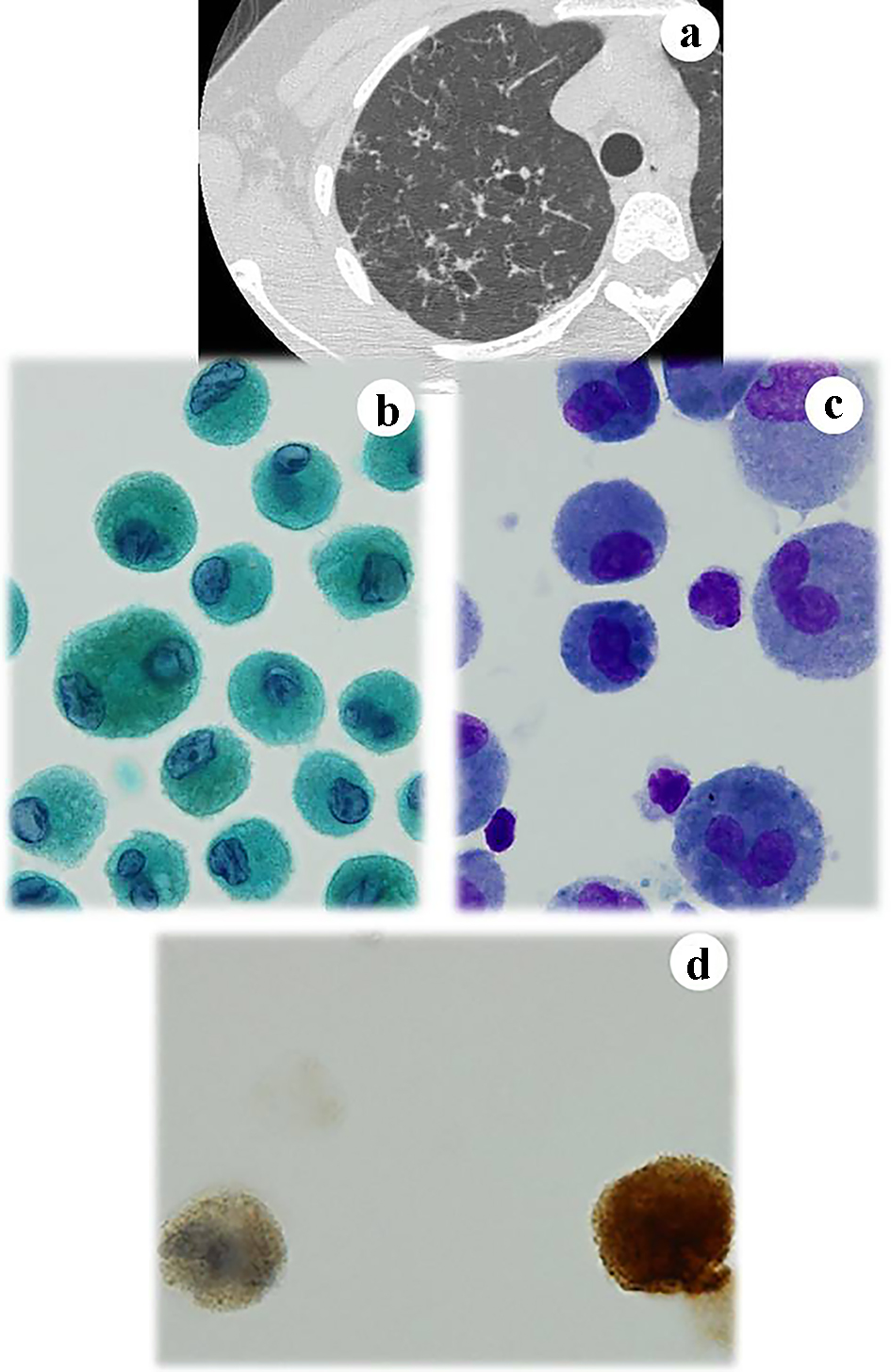 Click for large image | Figure 2. Case 2. CT of the lung reveals small cystic lesions in the right lung (a). A number of mononuclear and binuclear histiocytes are found in the BALF. They have (b) grooved nuclei and (c) prominent nucleoli. The cells are immunocytochemically positive for (d) S100. (b) Papanicolaou staining, × 400; (c) Giemsa staining, × 400; and (d) S100 immunocytochemistry, × 400. CT: computed tomography; BALF: bronchoalveolar lavage fluid. |
Case 3
In June 2014, a 33-year-old male noticed red pigmentation and surrounding erythema on his left dorsum manus. A dermoscopic examination revealed vasodilation in the lesion. A biopsy specimen showed granulomatous lesions consisting of atypical histiocytes, eosinophils, lymphocytes and neutrophils in the dermis (Fig. 3a). The histopathological diagnosis was skin LCH, and this was confirmed by immunohistochemical staining of S100 (Fig. 3b) and CD1a (Fig. 3c). The lesions also contained CD45RO-positive lymphocytes. However, they were immunohistochemically negative for the antibody of BRAFV600E antibodies. The patient’s skin lesions healed completely after 4 months of treatment with prednisolone.
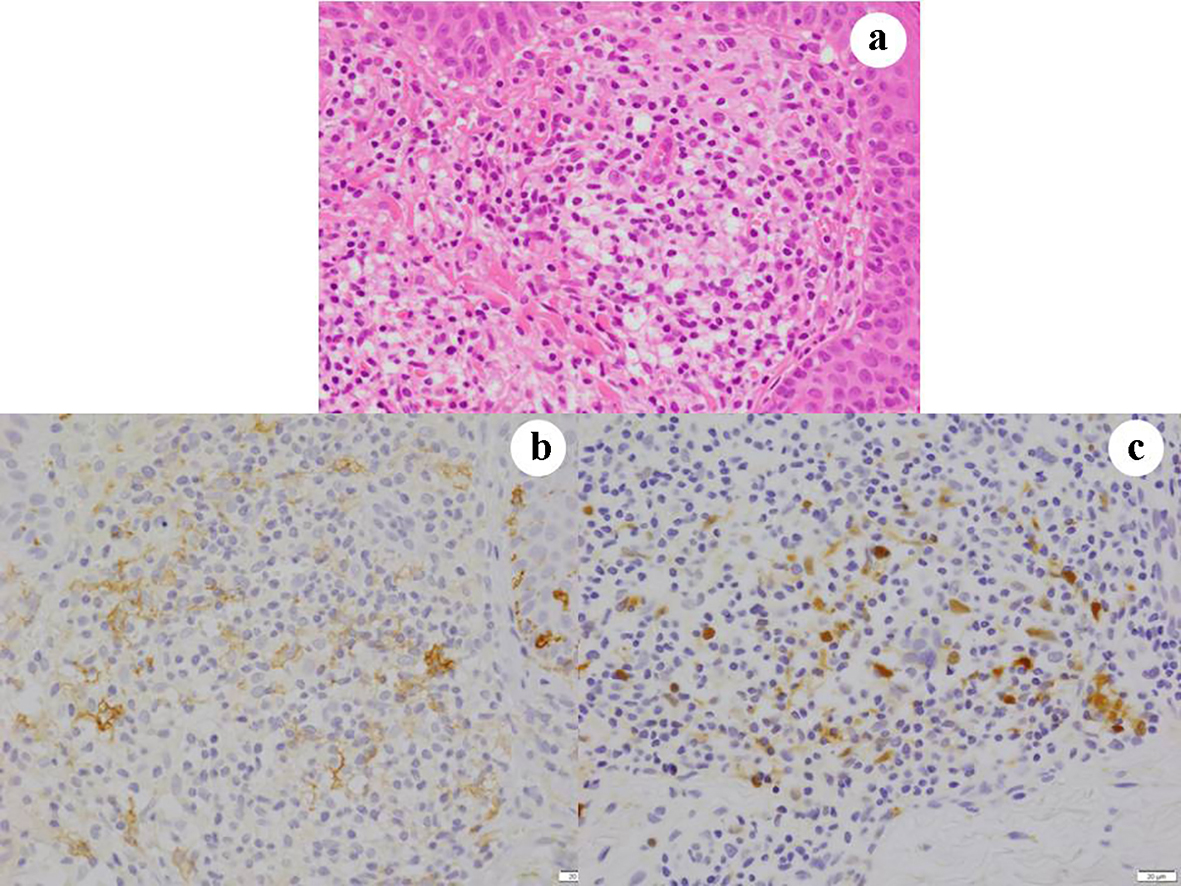 Click for large image | Figure 3. Case 3. Skin lesions consist of granulomatous lesions in the dermis (a). The lesions contain atypical histiocytes that are immunohistochemically positive for (b) CD1a and (c) S100. (a) Hematoxylin and eosin staining, × 400; (b) CD1a immunohistochemistry, × 400; and (c) S100 immunohistochemistry. |
Case 4
A 58-year-old male noticed erythroderma and edema on his left lower leg in November 2007. Enlargement of his bilateral inguinal lymph nodes was also indicated by a hematologyist. The stamp cytology of these lymph nodes, which were resected to make a pathological diagnosis, suggested LCH (Fig. 4a, b). Biopsy examination of these lymph nodes showed typical findings of LCH (Fig. 4c): atypical histiocytes with elongated nuclei possessing a coffee bean-like groove and slightly eosinophilic cytoplasm proliferated in conjunction with numerous eosinophils. Immunohistochemically, they were positive for CD1a (Fig. 4d) and S100 (Fig. 4e) antibodies. Immunohistochemically, atypical histiocytes were weakly positive for the BRAFV600E antibodies (Fig. 4f). Treatment with oral prednisolone resulted in the disappearance of the enlarged lymph nodes and erythroderma.
 Click for large image | Figure 4. Case 4. Stamp cytology of enlarged lymph nodes reveals that they contain numerous large histiocytes with irregular and grooved nuclei (a) accompanied with eosinophils (b). Histopathologically, the lesions consist of atypical histiocytes and are surrounded by eosinophils (c). Immunohistochemically, atypical histiocytes are positive for (d) CD1a and (e) S100. Some of the atypical histiocytes in the lesion are weakly positive for BRAFV600E. (a) Papanicolaou staining, × 400; (b) Giemsa staining, × 400; (c) Hematoxylin and eosin staining, × 400; (d) CD1a immunohistochemistry, × 400; (e) S100 immunohistochemistry; and (f) BRAFV600E immunohistochemistry. |
Case 5
A 2-year-old girl first presented to our hospital with intermittent claudication in July 2014. X-ray, MRI, CT and bone scan revealed small enhancing lesions in the bilateral iliac bones (Fig. 5a) and the parietal portion of the skull. An excisional biopsy was performed in both bones. A histopathological examination showed diffuse infiltration of histiocytes with destruction of the bony structure (Fig. 5b). In the lesion, LCs possessing intended, grooved or folded nuclei, a slightly eosinophilic cytoplasm and numerous eosinophils were noted. Immonohistochemical staining of CD1a (Fig. 5c) and S100 (Fig. 5d) confirmed the diagnosis of LCH in multiple bones (bilateral iliac bones and skull). Immunohistochemical staining of BRAFV600E was negative. She received prednisolone and the standard chemotherapy for LCH (Table 3), and remains asymptomatic at 4 years after the initiation of treatment.
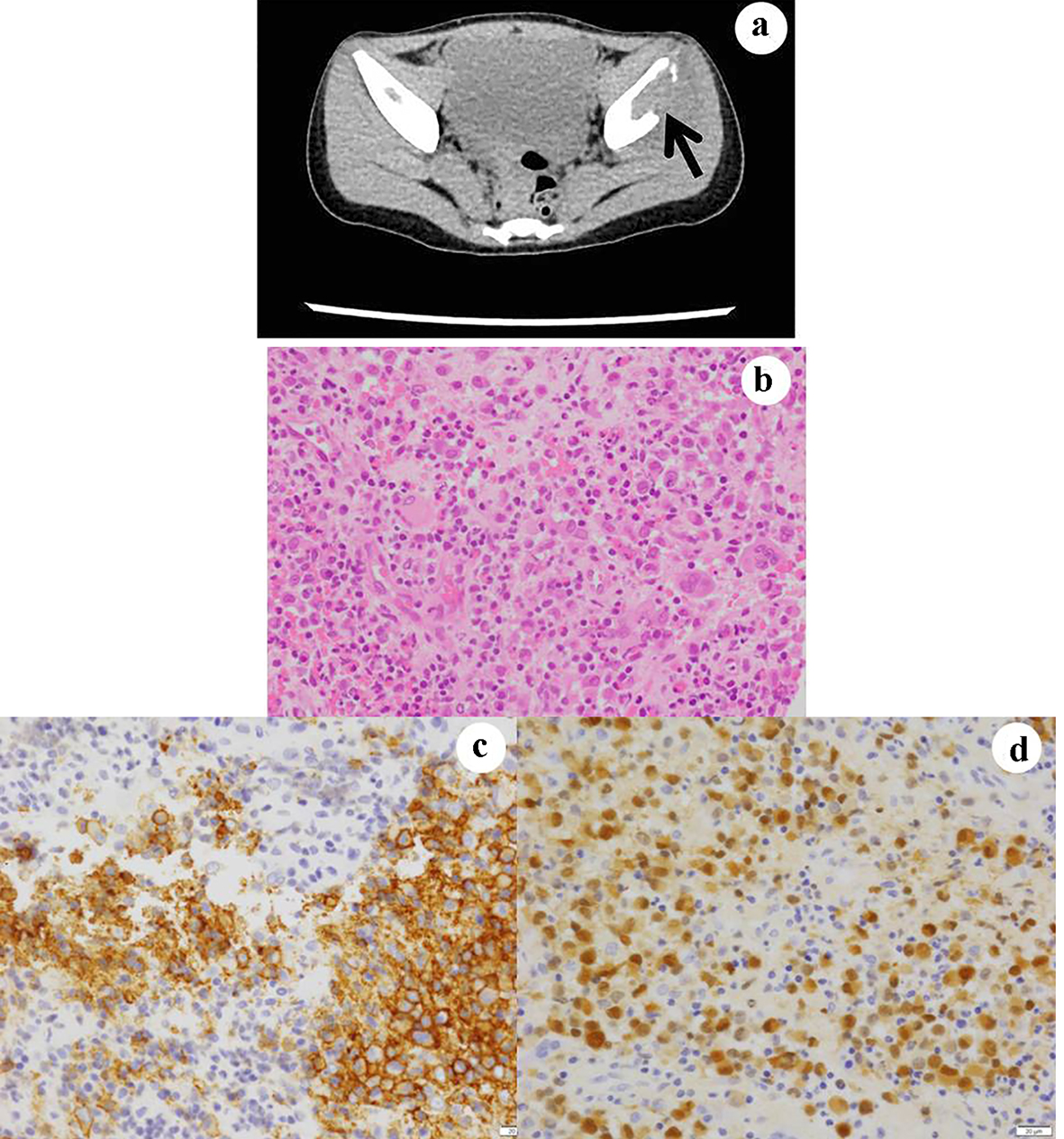 Click for large image | Figure 5. Case 5. The CT examination reveals osteolytic lesion in the iliac bones (a). In the biopsy specimens from the lesions, atypical and large histiocytes with single or multiple nuclei are present (b). They are immunohistochemically positive for (c) CD1a and (d) S100. (b) Hematoxylin and eosin stain, × 400; (c) CD1a immunohistochemistry, × 400; and (d) S100 immunohistochemistry. CT: computed tomography. |
Case 6
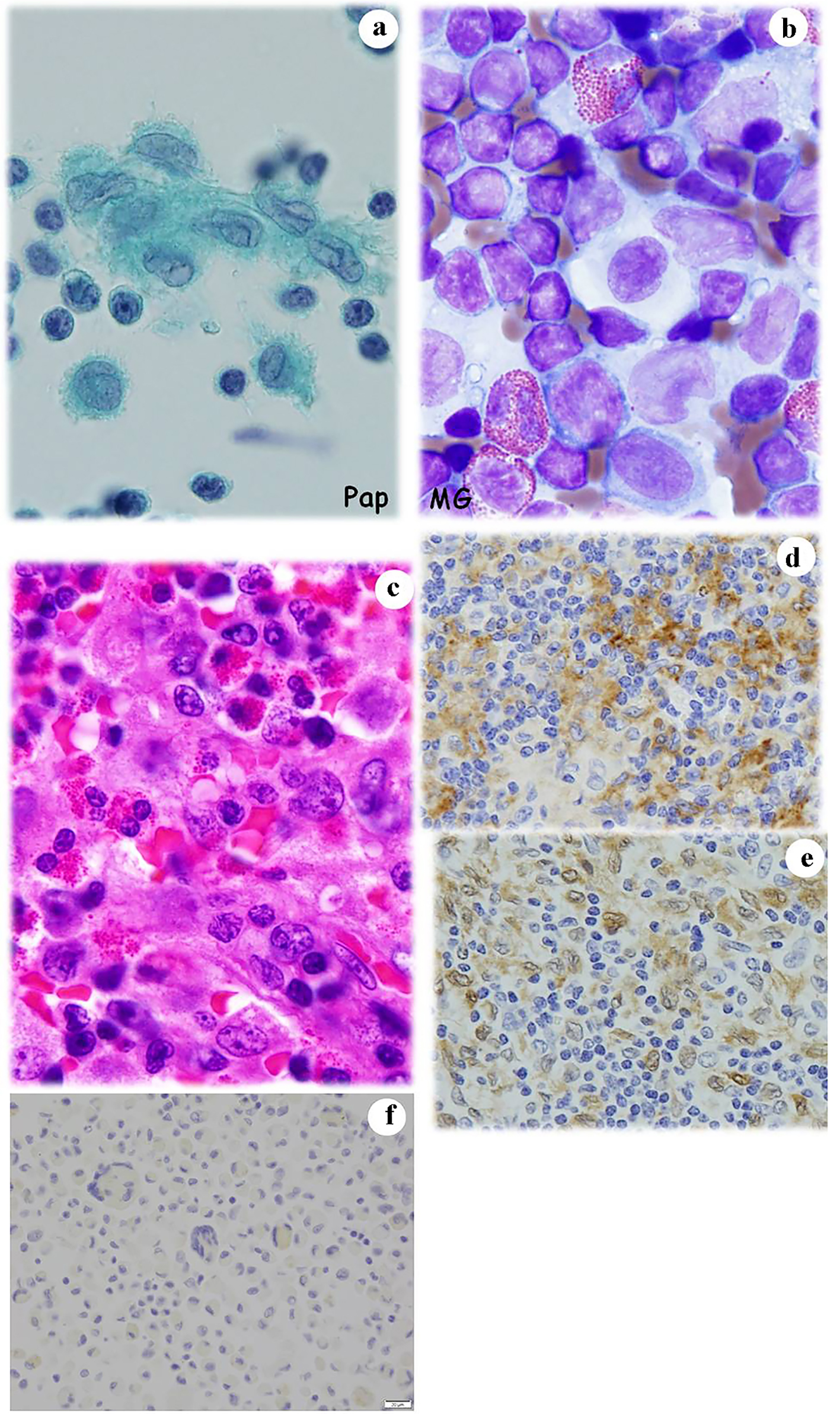
The patient was a 1-year-old girl who first presented to our hospital with fever and enlargement of the right cervical lymph node. In spite of the therapy against infectious lymphadenitis, she noticed intermittent pyrexia and could not stand on her own feet. At the same time, a lumbar spine X-ray examination revealed a compression fracture of the tertiary lumbar spine, calve platyspondyly (Fig. 6a), and punched-out lesions in the right femur and skull. CT-MRI revealed swellings of the lymph nodes of the right cervical region and bilateral peri-iliac arteries. Fine needle aspiration (FNA) cytology of cervical lymph node was performed to make an accurate diagnosis. The FNA cytology (Fig. 6b, c) and histopathology (Fig. 6d, e) findings suggested LCH based on the observation of LCs that were located centrally or eccentrically, with an oval or retiform shape, grooved or contorted nuclei, and an inflammatory background. Electron microscopy (EM) detected Birbeck’s granules (Fig. 6f) in the LCs and immunocytochemistry (Fig. 6g, h) and immunohistochemistry (Fig. 6i, j) were positive for S100 and CD1a. Based on these findings, a diagnosis of LCH was confirmed. However, immunohistochemical staining of BRAFV600E was negative. As in case 5, the patient was treated with prednisolone and received the standard chemotherapy for LCH (Table 3), and remains asymptomatic for 14 years.
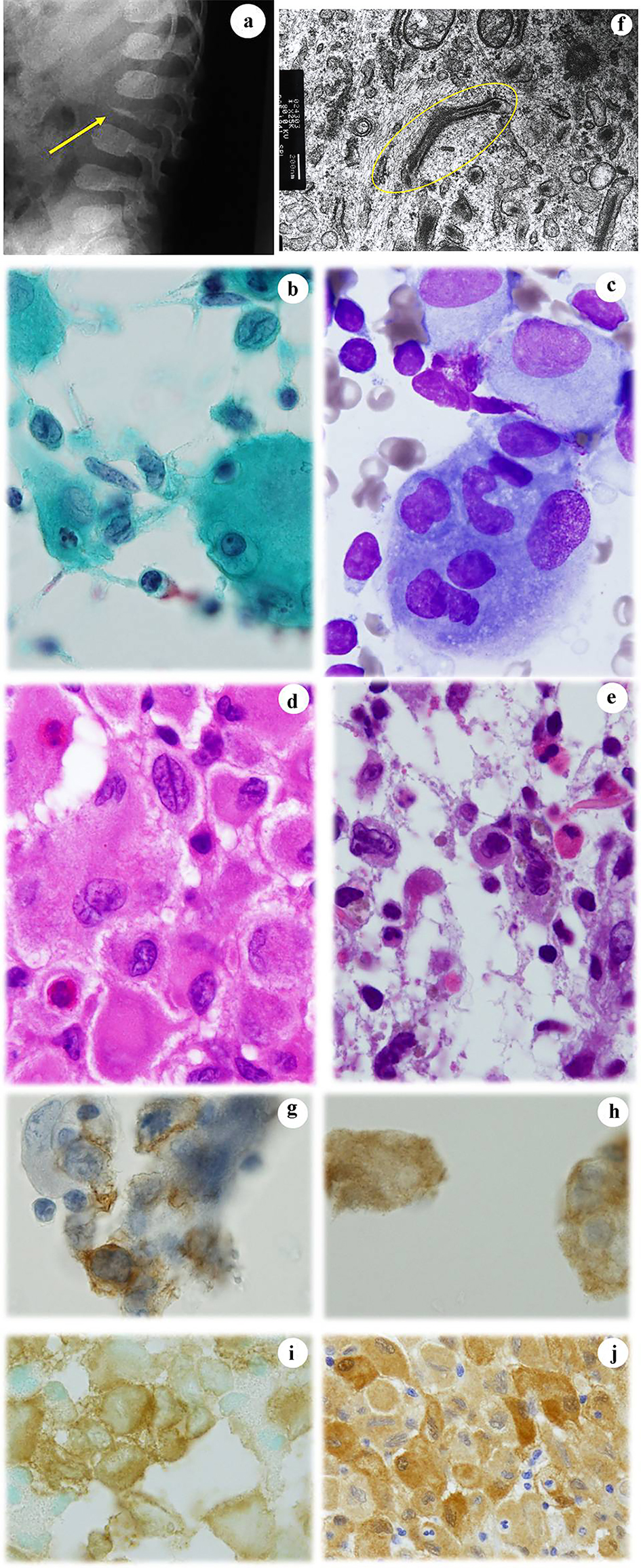 Click for large image | Figure 6. Case 6. (a) An X-ray examination shows platyspondyly of the lumbar calf (arrow). FNA cytology from the enlarged cervical lymph node shows a number of atypical and large histiocytes with single or multiple nuclei that have coffee bean-like grooves (b) or prominent nucleoli (c). Histopathologically, atypical and large histiocytes with grooves that are located together with eosinophils are noted in the biopsy specimens from (d) lymph node and (e) femur. EM examination reveals Birbeck’s granules (circled) in an atypical histiocyte (f). Atypical histiocytes are immunocytochemically positive for (g) CD1a and (h) S100. They are also immunohistochemically positive for (i) CD1a and (j) S100. (b) Papanicolaou staining, × 400; (c) Giemsa staining, × 400; (d) Hematoxylin and eosin staining, × 400; (e) Hematoxylin and eosin staining, × 400; (f) EM, × 62,500; (g) CD1a immunocytochemistry; (h) S100 immunocytochemistry, × 400; (i) CD1a immunohistochemistry, × 400; and (j) S100 immunohistochemistry. FNA: fine needle aspiration; EM: electron microscopy. |
Case 7
A 9-year-old girl visited our hospital in June 2010, to identify the cause of an erythematous nodule on the skin (right parietal portion), polydipsia and right cervical lymph nodes enlargement. X-ray, CT and MRI examinations suggested LCH involving multiple organs, including the skin, lungs (Fig. 7a), pituitary gland and lymph nodes. On skin biopsy specimens, granulomatous lesions were noted in the subepidermis (Fig. 7b). The lesions contained large tumor cells that were positive for CD1a (Fig. 7c) and S100 (Fig. 7d), and which aggregated in the perivascular region accompanied by eosinophils. We did not perform BRAFV600E immunohistochemistry. The patient remains free of symptoms at 8 years after chemotherapy.
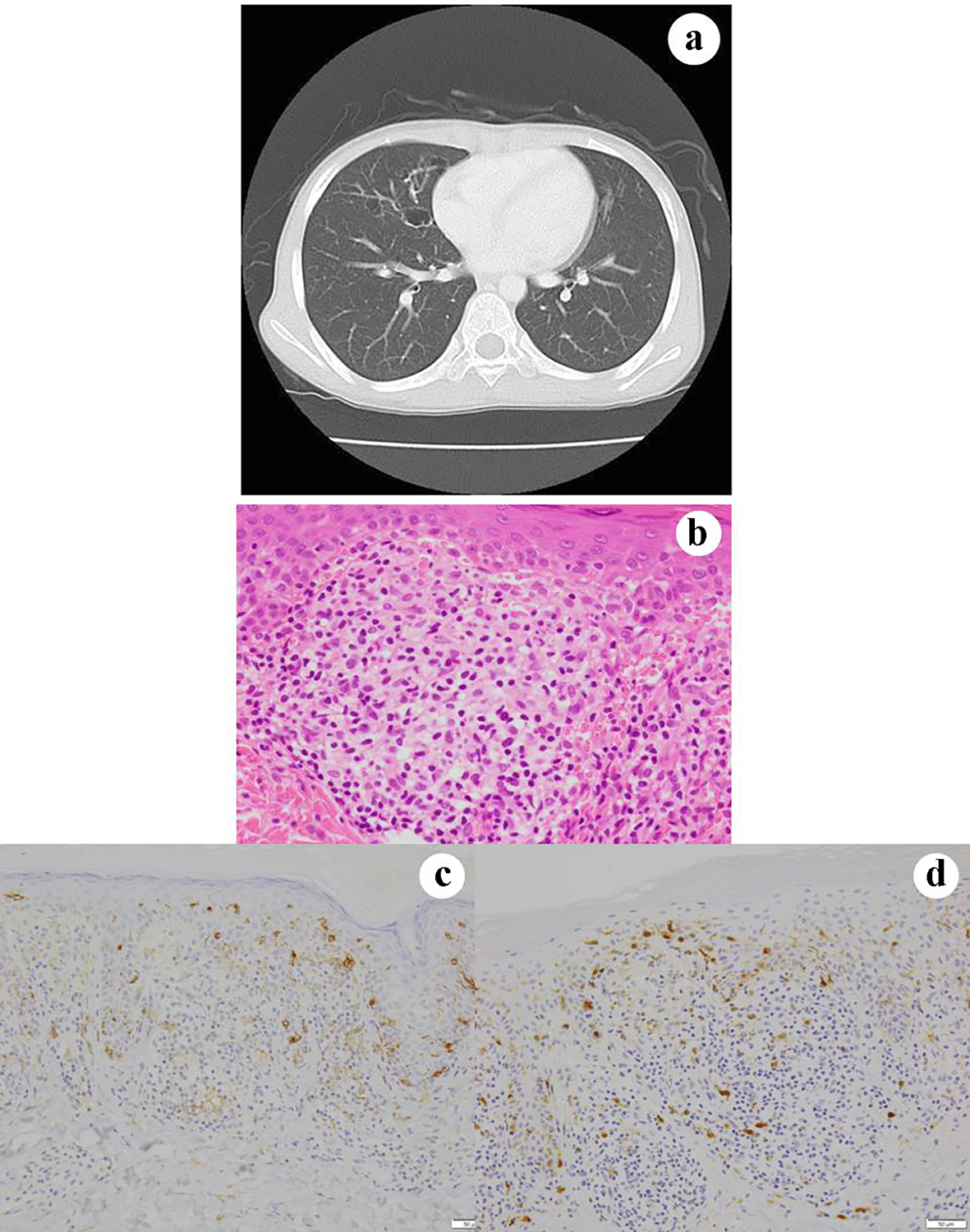 Click for large image | Figure 7. Case 7 (consultation case). Chest CT shows a cystic lesion in the right lung (a). Skin biopsy of the lesion shows granulomatous tissue in the dermis (b). CD1 positivity in the epidermis reveals the presence of native dendritic LCs. The lesion contains histiocyte-like cells that are weakly immunohistochemically positive for (c) CD1a and (d) S100. (b) Hematoxylin and eosin staining, × 400; (c) CD1a immunohistochemistry, × 400; and (d) S100 immunohistochemistry. CT: computed tomography; LCs: Langerhans cells. |
| Discussion | ▴Top |
We presented seven cases of LCH with or without involvement in several tissues, which were diagnosed and treated at Gifu Municipal Hospital. As reported previously, LCH developed in both sexes and at any age. Furthermore, CD1a- and S100-positive histiocyte-like cells were immunohistochemically and/or immunocytochemically confirmed in the hitopathological or cytological specimens from the lesions, while Birbeck’s granules were detected by electron microscopy in only one case (case 6). In all cases with the exception of case 3, where the LCH lesion was limited to the skin, imaging was helpful for suspecting LCH; these sites included the lung, lymph nodes, bone and central nervous system (CNS). In one case (case 2), pulmonary LCH was suspected on a pulmonary CT examination performed in a health check and was diagnosed by BALF cytology, which detected CD1a-positive histiocyte-like cells. LCH can be easily diagnosed by FNA cytology based on its morphological and immunocytochemical characteristics [15]. While familial clustering as well as chromosomal instability is implicated in disease occurrence [8, 9], we did not notice such cases. Oncogenic BRAFV600E mutations are reported in almost half or more than half of LCH cases [9]; however, these mutations were detected by immunohistochemistry in just two (28.6%) of our cases (cases 1 and 4).
LCH is known for its heterogenous clinical presentation, which ranges mild, self-healing (by cessation of smoking) disease to more severe cases involving risk organs [16] (defined by the presence of at least one of the following: hematopoietic system (bicytopenia or pancytopenia), liver (hepatomegaly and/or dysfunction) and/or spleen (splenomegaly)), and which sometimes involves the CNS with mass lesions, pituitary lesions resulting in diabetes insipidus (DI) as found in case 7 and/or a neurodegenerative syndrome [4]. Risk of DI development was decreased in patients with mastoid, temporal, orbital and skull base bone lesions when they received systemic chemotherapy [17]. Localized LCH, defined as single-system LCH (SS-LCH), often affects the bone, skin or lymph nodes, as was observed in cases 2, 3 and 4. SS-LCH is a low-risk disease and usually results in spontaneous remission and a favorable outcome [9]. As experienced in case 3, LCH skin lesions may appear as scaly and greasy rashes, small and solid reddish papules, purplish-red spots, or ulcerations with a small abscess.
In contrast to LCH in children, adult LCH most commonly develops in the lung, followed by the bone and skin with involvement of the genital and oral mucosa and nails [9]. Pulmonary LCH is characterized by micronodules and/or honeycomb lesions [3, 18]. Although we did not notice adult genital, oral or nail LCH in this series, cases 1 and 2 were involved young adults with pulmonary LCH. Both cases were associated with smoking and the lesions disappeared completely with smoking cessation [4, 7, 9]. However, when pulmonary LCH occurs as a part of multi-system LCH (MS-LCH) in a child, chemotherapy is required, as was experienced in case 7 [4, 7, 9]. MS-LCH is associated with high morbidity and resistance to treatment, with recurrent disease developing in 20% of patients [4, 7, 9].
Skeletal LCH in adults usually affects the skull, ribs and long bones; clinical manifestations include pain or spontaneous fractures [9]. Patients with a new onset of diabetes insipidus should be evaluated due to the possibility of LCH involving the pituitary gland [9]. Other endocrine LCH should be considered in adults when thyroid dysfunction and ovarian failure are found.
The clinical classification of LCH is based on empirical results [3, 7, 9]: patients presenting with the involvement of one organ, namely SS-LCH have an excellent prognosis, while patients with the involvement of two or more organs, namely MS-LCH, particularly the liver and/or hematopoietic system may show progressive disease, which is associated with considerable mortality.
In conclusion, LCH is a rare disease with a wide range of clinical manifestations that may involve several types of tissue. Thus, an early diagnosis and appropriate therapy is needed for the appropriate treatment of the disease. In this context, the discovery of BRAFV600E and MAP2K1 mutations can be used to assess the patient’s degree of risk and is relevant for new treatment strategies against the disease. In the current series, prolonged disease control was achieved in children with MS-LCH (cases 4, 5 and 7) using MTX-based chemotherapy. Due to the rarity of this disease, reports of a small patient series may provide additional data that are valuable for therapy. However, multicenter studies with international collaboration will be necessary for the development of rational therapeutic strategies that improve patient outcomes.
Acknowledgments
The authors would like to thank Toshie Naraki for her assistance in manuscript review process.
Financial Disclosure
This work was not supported by any funding agency.
Conflict of Interest
The authors have no conflict of interest.
Informed Consent
All patients or their parents provided their written informed consent.
Author Contributions
TT designed and performed study, analyzed data and wrote the paper. RN and RA performed immunocytochemistry and immunohistochemistry. FE, AO, NK, TS, RM and MM performed cytological diagnosis. NW performed histopathological diagnosis.
| References | ▴Top |
- Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, Requena-Caballero L, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127(22):2672-2681.
doi pubmed - Hand A. Polyuria and tuberculosis. Arch. Pediatr. 1893;10:673-675.
- Harmon CM, Brown N. Langerhans cell histiocytosis: a clinicopathologic review and molecular pathogenetic update. Arch Pathol Lab Med. 2015;139(10):1211-1214.
doi pubmed - Haroche J, Cohen-Aubart F, Rollins BJ, Donadieu J, Charlotte F, Idbaih A, Vaglio A, et al. Histiocytoses: emerging neoplasia behind inflammation. Lancet Oncol. 2017;18(2):e113-e125.
doi - Howarth DM, Gilchrist GS, Mullan BP, Wiseman GA, Edmonson JH, Schomberg PJ. Langerhans cell histiocytosis: diagnosis, natural history, management, and outcome. Cancer. 1999;85(10):2278-2290.
doi - Grana N. Langerhans cell histiocytosis. Cancer Control. 2014;21(4):328-334.
doi pubmed - Kobayashi M, Tojo A. Langerhans cell histiocytosis in adults: Advances in pathophysiology and treatment. Cancer Sci. 2018;109(12):3707-3713.
doi pubmed - Durham BH. Molecular characterization of the histiocytoses: Neoplasia of dendritic cells and macrophages. Semin Cell Dev Biol. 2019;86:62-76.
doi pubmed - Hutter C, Minkov M. Insights into the pathogenesis of Langerhans cell histiocytosis: the development of targeted therapies. Immunotargets Ther. 2016;5:81-91.
doi pubmed - Morimoto A, Ikushima S, Kinugawa N, Ishii E, Kohdera U, Sako M, Fujimoto J, et al. Improved outcome in the treatment of pediatric multifocal Langerhans cell histiocytosis: results from the Japan Langerhans Cell Histiocytosis Study Group-96 protocol study. Cancer. 2006;107(3):613-619.
doi pubmed - Tran G, Huynh TN, Paller AS. Langerhans cell histiocytosis: A neoplastic disorder driven by Ras-ERK pathway mutations. J Am Acad Dermatol. 2018;78(3):579-590 e574.
doi pubmed - Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: History, classification, pathobiology, clinical manifestations, and prognosis. J Am Acad Dermatol. 2018;78(6):1035-1044.
doi pubmed - Allen CE, Merad M, McClain KL. Langerhans-cell histiocytosis. N Engl J Med. 2018;379(9):856-868.
doi pubmed - Ballester LY, Cantu MD, Lim KPH, Sarabia SF, Ferguson LS, Renee Webb C, Allen CE, et al. The use of BRAF V600E mutation-specific immunohistochemistry in pediatric Langerhans cell histiocytosis. Hematol Oncol. 2018;36(1):307-315.
doi pubmed - Hang JF, Siddiqui MT, Ali SZ. Fine needle aspiration of Langerhans cell histiocytosis: a cytopathologic study of 37 cases. Acta Cytol. 2017;61(2):96-102.
doi pubmed - Lahey ME. Prognostic factors in histiocytosis X. Am J Pediatr Hematol Oncol. 1981;3(1):57-60.
- Grois N, Potschger U, Prosch H, Minkov M, Arico M, Braier J, Henter JI, et al. Risk factors for diabetes insipidus in langerhans cell histiocytosis. Pediatr Blood Cancer. 2006;46(2):228-233.
doi pubmed - Roden AC, Yi ES. Pulmonary Langerhans cell histiocytosis: an update from the pathologists' perspective. Arch Pathol Lab Med. 2016;140(3):230-240.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.