

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 11, Number 4, April 2020, pages 90-93
Unexpected Genotype in a Non-Transfusion Dependent Thalassemia Family
Cristina Curcioa, d, Valentina Giannonea, Elena Benzonia, Claudia Cesarettib, Elena Cassinerioc, Manuela Seiaa, Giovanna Graziadeic
aMedical Genetics Laboratory, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, via Francesco Sforza 35, 20122 Milan, Italy
bMedical Genetics Unit Woman, Child and Newborn Department, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, via Francesco Sforza 35, 20122 Milan, Italy
cRare Diseases Center, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, via Francesco Sforza 35, 20122 Milan, Italy
dCorresponding Author: Cristina Curcio, Medical Genetics Laboratory, IRCCS Ca’ Granda Foundation, via Francesco Sforza 35, 20122 Milan, Italy
Manuscript submitted July 11, 2019, accepted January 15, 2020
Short title: Unexpected Genotype in an NTDT Family
doi: https://doi.org/10.14740/jmc3335
| Abstract | ▴Top |
Non-transfusion dependent thalassemia (NTDT) is an inherited hemoglobin disorder characterized by an α/non-α globin chain imbalance of variable severity, resulting in a wide spectrum of clinical manifestations. The coinheritance of additional α genes with a beta-thalassemia heterozygous mutation has a well-known negative effect. Triplication or quadruplication alone are mostly found by chance, but the coinheritance with β mutations can worsen the very mild anemia to a more severe hematological and clinical phenotype causing NTDT, depending on the severity of beta mutations. We describe a case of a 38-year-old β-thalassemia trait, pregnant woman at 33 weeks of gestation with supernumerary α-globin genes and two β-globin defects.
Keywords: Non-transfusion dependent thalassemia; Supernumerary α-globin genes; α and β-globin defects coinheritance; Genetic counselling
| Introduction | ▴Top |
Non-transfusion dependent thalassemia (NTDT) [1] is an inherited hemoglobin disorder characterized by an α/non-α globin chain imbalance of variable severity, resulting in a wide spectrum of clinical manifestations. Phenotype reflects the genetic heterogeneity which depends on different molecular mechanisms, the most important of which are: 1) The coinheritance of two β-globin defects (homozygous or compound heterozygous for mild β+ mutations or compound heterozygous for β0 and β+ mutations) with relative excess of unmatched alpha chains [2, 3]; 2) The imbalance of α/non-α globin-chain synthesis ratio due to the coinheritance of a β mutation with supernumerary α-globin genes, resulting in an absolute excess of unmatched α chains [4-8]. Excess α-globin chains precipitate damaging erythroid membrane structures and increase destruction of erythroid cells by peripheral hemolysis and premature destruction of precursors in the bone marrow, resulting in anemia and ineffective erythropoiesis [2]. The mechanism of α-globin gene copy number alterations is based on a misalignment and reciprocal crossover involving α-globin gene cluster, due to the high homology of HBA1 and HBA2 genes. This recombinant event generates both alleles with a common α deletion (3.7 kb I, II, III, 4.2 kb) and alleles with three active α-globin genes (αααanti-α3.7; αααanti-α4.2) [8].
A second unequal crossover within a normal chromosome and a chromosome bearing the triplication, causes the quadruplication (ααααanti-α3.7; ααααanti-α4.2) of α-globin genes [9]. During evolution these rearranged chromosomes undergo positive selection in the population [10]. The negative effect of additional α genes coinheritance with a beta-thalassemia heterozygous mutation is well known [2, 11].
| Case Report | ▴Top |
We describe a case of a 38-year-old β-thalassemia trait, pregnant woman at 33 weeks of gestation referred to the Rare Diseases Centre of Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy, for severe anemia requiring blood transfusion support.
Her anamnesis was not clinically relevant before pregnancy; she referred an average hemoglobin (Hb) level of 9.5 g/dL; the clinical picture showed normal spleen and liver size.
To better explain the worsening of Hb level during pregnancy, hematological and hemoglobin patterns were assessed, showing Hb 7.2 g/dL, red blood cell (RBC) 3.44 × 106/mm3, mean corpuscular volume (MCV) 70.1 fL, HBA2 4.9%, HbF 3.8% (high-performance liquid chromatography (HPLC) Biorad D100, Biorad Laboratories, Hercules, CA, USA), and globin genes molecular analysis was performed (BigDye Terminator Cycle Sequencing Ready Reaction Kit v.1.1).
DNA sequencing of β-globin gene revealed β+ IVS-I-110 (c.93-21G>A) and β++/+ IVS-II-726 (c.316-125A>G) mutations; the second mutation is very rare and was reported in Database of Human Hemoglobin Variants and Thalassemia mutations (HbVar Database http://globin.bx.psu.edu/hbvar/menu.html) both as β+ and βsilent thalassemia mutation [12, 13]. HBB gene analysis was inconclusive to solve the proband clinical picture. An α-globin genes cluster study with multiplex ligation-dependent probe amplification (MLPA) method was performed according to the manufacturer’s instructions (SALSA MLPA kit P140B2 HBA; MRC-Holland, Amsterdam, The Netherlands). MLPA probe set resulted in a complex rearrangement of HBA1 and HBA2 gene spanning from probe HBA2-up (373) to HBA1-intr.2 (165) (Fig. 1) with a different ratio which could be interpreted as a combination of ααααanti-α4.2 quadruplication and αααanti-α3.7 triplication, resulting in seven active genes.
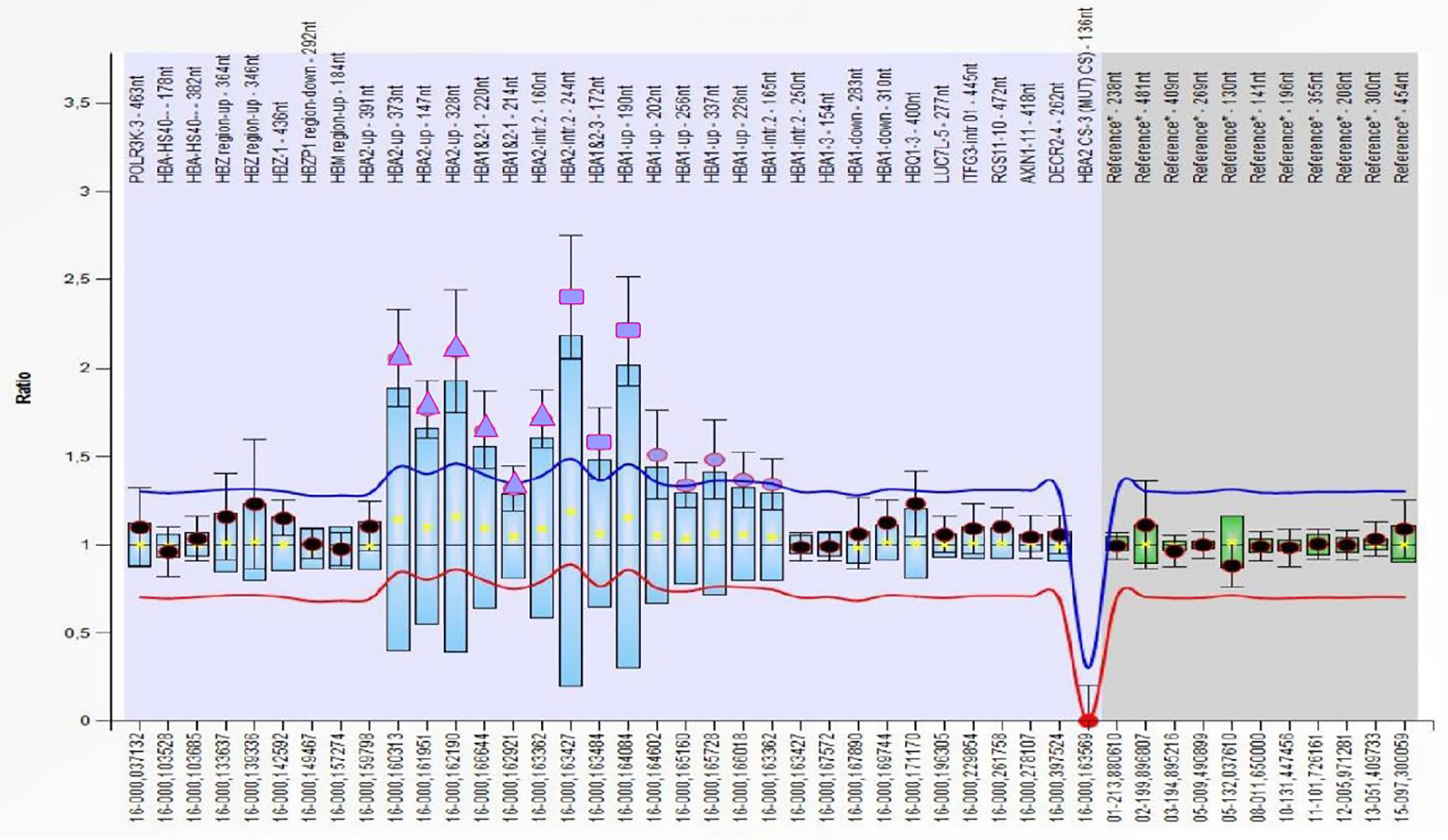 Click for large image | Figure 1. Profile of MLPA probemix P140 HBA cluster of the proband carrying a multiplication of HBA genes. The triangles represent the probe ratio of about 2 corresponding to alpha genes quadruplication; the dots represent the probe ratios of about 1.5 corresponding to alpha genes triplication, while the squares correspond to overlapping probes. MLPA: multiplex ligation-dependent probe amplification. |
To study the allelic segregations, family hematological and hemoglobin pattern were investigated (Table 1). The mother and brother’s hematological phenotype was consistent with β-thalassemia carrier, while the father’s was normal. Besides the patient, only the mother showed slightly higher HbF levels, resulting in heterozygosity of -158Gγ (C→T) Xmn1 polymorphism detected by sequencing analysis of gamma globin genes; the same alteration was found in the proband. Even if the HbF level was normal in the father and the brother, gamma-globin gene analysis was performed; the c.227T>C in HBG1 globin gene (HbF-Sardinia) was identified only in the father, known to be associated with increase of 10% of the total HbF.
 Click to view | Table 1. Comparison Between Proband’s and Relatives’ Hematological and HPLC Data |
HBB direct sequencing and MLPA analysis of the α-globin gene cluster revealed that the mother was a carrier of β+ IVS-I-110 associated with a ααααanti-α4.2 quadruplication, while the father was a carrier of β++ IVS-II-726 and αααanti-α3.7 triplication. Molecular analysis was also extended to the proband’s brother and showed the presence of β+ IVS-I-110 in heterozygous state and αααanti-α3.7 triplication. All the relatives were asymptomatic.
Triplication or quadruplication alone are mostly found by chance, but the coinheritance with β mutations could worsen the very mild anemia to a more severe hematological and clinical phenotype causing NTDT, depending on the severity of beta mutations [4]. Although HbF is usually slightly increased in these molecular patterns, we underline that in this case HbF levels are not significant and are sustained by gamma-globin genes alterations.
In the proband’s partner HBB gene was studied by sequencing analysis and HBA cluster by MLPA technique, resulting both negative.
| Discussion | ▴Top |
To define reproductive risk in these cases, hematological screening and molecular analysis of beta-globin mutations and α-globin genes rearrangement are strongly recommended in the partner, even if he has a normal hematologic profile, to exclude the presence of genetic alterations potentially worsening offspring’s hematologic phenotype. Although the partner had no evidence of hemoglobin disorder, the couple still had a reproductive risk of having offspring with NTDT: specifically all their children would inherit one β mutation in association with supernumerary α genes. Therefore, the clinical picture of the offspring could vary from a very mild form, likely asymptomatic, deriving from the coinheritance of β++/+ IVS-II-726 and αααanti-α3.7 triplication to a mild/moderate form, if the child inherits the β+ IVS-I-110 with ααααanti-α4.2 quadruplication. In this molecular pattern, even if the reproductive risk is 100%, all the possible genotype combinations result generally in mild form of NTDT, without major or early onset complications.
Acknowledgments
The authors would like to acknowledge Fondazione G. e D. De Marchi Onlus, Milan, Italy for supporting to this work.
Financial Disclosure
None to declare.
Conflict of Interest
The authors declare no conflict of interest.
Informed Consent
All subjects signed the informed consent form before blood sampling.
Author Contributions
C. Curcio and G. Graziadei analyzed the data and wrote the paper. V. Giannone and E. Benzoni performed molecular analysis and interpretation of data. M. Seia contributed in proofreading and language editing. C. Cesaretti performed genetic counselling. E. Cassinerio performed evaluation of hematological and biochemical parameters.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Weatherall DJ. The definition and epidemiology of non-transfusion-dependent thalassemia. Blood Rev. 2012;26(Suppl 1):S3-6.
doi - Harteveld CL, Refaldi C, Cassinerio E, Cappellini MD, Giordano PC. Segmental duplications involving the alpha-globin gene cluster are causing beta-thalassemia intermedia phenotypes in beta-thalassemia heterozygous patients. Blood Cells Mol Dis. 2008;40(3):312-316.
doi pubmed - Sullivan KM, Anasetti C, Horowitz M, Rowlings PA, Petersdorf EW, Martin PJ, Clift RA, et al. Unrelated and HLA-nonidentical related donor marrow transplantation for thalassemia and leukemia. A combined report from the seattle marrow transplant team and the international bone marrow transplant registry. Ann N Y Acad Sci. 1998;850:312-324.
doi pubmed - Liu S, Jiang H, Wu MY, Zhang YL, Li DZ. Thalassemia intermedia caused by 16p13.3 sectional duplication in a beta-thalassemia heterozygous child. Pediatr Hematol Oncol. 2015;32(5):349-353.
doi pubmed - Wainscoat JS, Kanavakis E, Wood WG, Letsky EA, Huehns ER, Marsh GW, Higgs DR, et al. Thalassaemia intermedia in Cyprus: the interaction of alpha and beta thalassaemia. Br J Haematol. 1983;53(3):411-416.
doi pubmed - Thein SL, Hesketh C, Wallace RB, Weatherall DJ. The molecular basis of thalassaemia major and thalassaemia intermedia in Asian Indians: application to prenatal diagnosis. Br J Haematol. 1988;70(2):225-231.
doi pubmed - Beris P, Solenthaler M, Deutsch S, Darbellay R, Tobler A, Bochaton-Pialat ML, Gabbiani G. Severe inclusion body beta-thalassaemia with haemolysis in a patient double heterozygous for beta(0)-thalassaemia and quadruplicated alpha-globin gene arrangement of the anti-4.2 type. Br J Haematol. 1999;105(4):1074-1080.
doi pubmed - Sollaino MC, Paglietti ME, Perseu L, Giagu N, Loi D, Galanello R. Association of alpha globin gene quadruplication and heterozygous beta thalassemia in patients with thalassemia intermedia. Haematologica. 2009;94(10):1445-1448.
doi pubmed - Gu YC, Landman H, Huisman TH. Two different quadruplicated alpha globin gene arrangements. Br J Haematol. 1987;66(2):245-250.
doi pubmed - Higgs DR, Hill AV, Bowden DK, Weatherall DJ, Clegg JB. Independent recombination events between the duplicated human alpha globin genes; implications for their concerted evolution. Nucleic Acids Res. 1984;12(18):6965-6977.
doi pubmed - Graziadei G, Refaldi C, Barcellini W, Cesaretti C, Cassinero E, Musallam KM, Cappellini MD. Does absolute excess of alpha chains compromise the benefit of splenectomy in patients with thalassemia intermedia? Haematologica. 2012;97(1):151-153.
doi pubmed - Patrinos GP, Giardine B, Riemer C, Miller W, Chui DH, Anagnou NP, Wajcman H, et al. Improvements in the HbVar database of human hemoglobin variants and thalassemia mutations for population and sequence variation studies. Nucleic Acids Res. 2004;32(Database issue):D537-541.
doi pubmed - Agouti I, Bennani M, Nezri M, Levy N, Badens C. Beta-thalassemia intermedia due to two novel mutations in the promoter region of the beta-globin gene. Eur J Haematol. 2008;80(4):346-350.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.