

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 10, Number 12, December 2019, pages 348-353
Acute Tubulointerstitial Nephritis as a Sole Manifestation of Immunoglobulin G4-Related Disease
Manaf Aljishia, b, e, Zaw Theta, Thin Hanc, Krishan Madhand
aRenal Department, Rockhampton Hospital, Queensland, Australia
bUniversity of Queensland, St Lucia QLD 4072, Australia
cMedicine and Nephrology, Rockhampton Hospital, Rockhampton QLD 4700, Australia
dRenal Medicine Department, Hervey Bay Hospital, Queensland, Australia
eCorresponding Author: Manaf Aljishi, Renal Department, Rockhampton Hospital, Queensland, Australia
Manuscript submitted December 1, 2019, accepted December 10, 2019
Short title: IgG4-RD Presenting as Renal Failure
doi: https://doi.org/10.14740/jmc3395
| Abstract | ▴Top |
This is a case report of a patient who developed severe acute kidney disease with kidney biopsy showing interstitial nephritis, plasma cell infiltration and immunoglobulin G4 (IgG4) expression consistent with IgG4-related kidney disease. There were no other systemic features of IgG4-related disease. The patient was treated with corticosteroids and mycophenolate. This case highlights the need to consider IgG4-related kidney disease even in the absence of other systemic features. Isolated renal involvement is underrecognized and can lead to missed diagnosis. It also illustrates that mycophenolate can be used as a steroid sparing agent in this condition; an observation that adds to the limited literature in this field.
Keywords: Acute renal failure; IgG4; IgG4-related disease; IgG4-related kidney disease; Tubulointerstitial nephritis
| Introduction | ▴Top |
Immunoglobulin G4 (IgG4)-related disease (RD) is a systemic inflammatory disorder which presents as inflammatory enlarging lesions. IgG4 disease has been described in nearly every organ system, and the constellation of location and behaviour of the multiple organ involvements often provide clues to the diagnosis. However, single organ involvement is more difficult to diagnose, and this delay in diagnosis leads to significant risk of organ damage. This is especially the case if the organ involved is the kidney, which is not often recognized. Renal involvement can present in various insidious ways and thus a high index of suspicion is required to diagnose especially in cases lacking systemic features. Knowledge about unusual sole manifestation of such uncommon condition is important for prompt diagnosis and proper management. We describe a patient who presented with renal failure as the only manifestation of IgG4-RD.
| Case Report | ▴Top |
A 63-year-old man presented in October 2017 with fatigue and non-specific tiredness. There were no infective symptoms, nausea, abdominal pain, rash, join pains or urinary symptoms apart from longstanding nocturia treated with oxybutynin 2.5 mg twice daily. He had a known diagnosis of Parkinson’s disease managed with extended-release pramipexole 3 mg daily and levodopa/benserazide 100 mg/25 mg daily. He had not taken any non-prescribed medications. General physical examination was normal, and he appeared euvolaemic. Blood pressure was 160/93 mm Hg.
Investigations
Laboratory testing showed acute renal failure with creatinine 508 µmol/L, estimated glomerular filtration rate (eGFR) 10 mL/min/1.73m2 (previous eGFR was 50 mL/min/1.73m2 in January 2016), potassium 5.4 mmol/L, bicarbonate 16 mmol/L, phosphate 2.32 mmol/L, corrected calcium 2.19 mmol/L, albumin 36 g/L, hemoglobin 112 g/L, platelet count 440 × 109/L, and white cell count 8.6 × 109/L.
Urine analysis showed 30 × 106 leucocytes/L and 30 × 106 erythrocytes/L, with no dysmorphic erythrocytes or casts. There was proteinuria of 1,200 mg/24 h. Urine culture showed no growth. Kidney ultrasound showed normal-sized kidneys with normal parenchyma, non-obstructive calculi in the right kidney and no hydronephrosis. Abdominal computed tomography (CT) scan revealed no obstructive uropathy, lymphadenopathy, masses or infective source.
Proteinase-3 (PR3) and myeloperoxidase (MPO) anti-neutrophil cytoplasmic autoantibody (ANCA) were negative. Antinuclear antibody (ANA) titre was 1:40, and extractable nuclear antigen and anti-double stranded DNA were negative. Serum immunoglobulins were normal except elevated IgG of 27 g/L (normal range: 7.0 - 16.0 g/L). C3 and C4 complement levels were low (0.83 g/L (0.90 - 1.80) and 0.07 g/L (0.10 - 0.40), respectively). The remainder of the immunology panel, including hepatitis B, hepatitis C, human immunodeficiency virus (HIV), anti-glomerular basement membrane, serum protein electrophoresis and immunofixation, serum free light chains, urine electrophoresis and immunofixation, were normal.
Kidney biopsy findings and diagnosis
He proceeded to have a kidney biopsy which demonstrated periglomerular fibrosis with no glomerular hypercellularity, segmental sclerosis or crescent formation. There was no evidence of arteriolar hyalinosis or vasculitis. There was extensive, dense lymphocytic infiltration involving the renal capsule, the cortex and the outer medulla with severe chronic tubulointerstitial nephritis (TIN) with marked tubular atrophy. No granulomata were identified, and there was no significant neutrophilia. Plasma cell-rich TIN with increased IgG4-plasma cells was demonstrated on immunohistochemistry. Mott cells were also seen. These are atypical plasma cells characterized by an accumulation of multiple Russell bodies, globular cytoplasmic inclusions composed of immunoglobulin that have not been released. On immunofluorescence, tissue showed granular tubular reactivity for IgG, kappa and lambda light chains, weak IgM reactivity, trace C3 reactivity, and no IgA or C1q reactivity (Fig. 1).
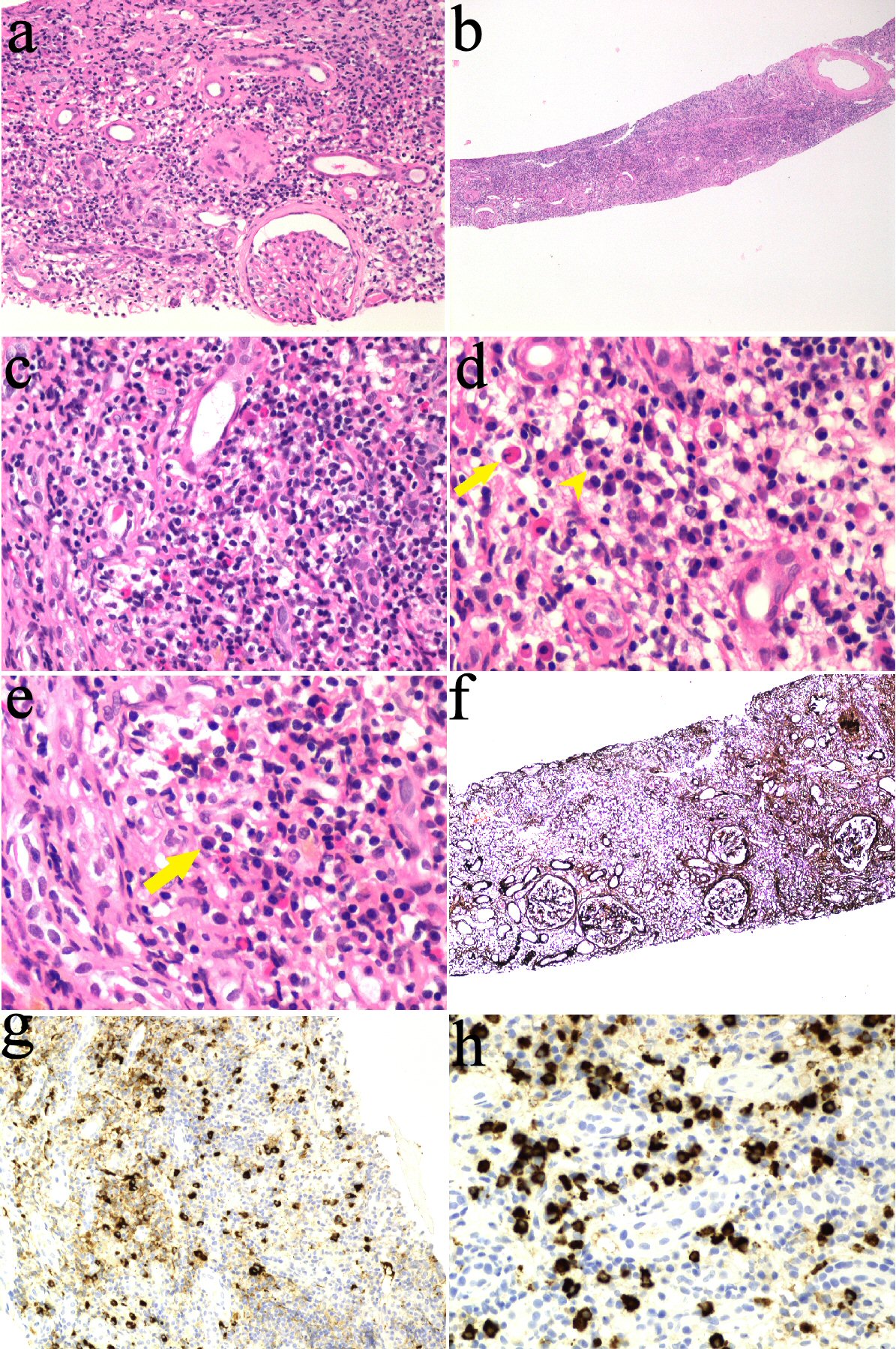 Click for large image | Figure 1. kidney biopsy findings. (a, b) Interstitial inflammation on hematoxylin and eosin (H&E) stain. (c) Lymphocytes, plasma cells and eosinophils. (d) Plasma (arrowhead) and Mott cells (arrow). (e) Plasma cell (arrow). (f) Marked tubular atrophy on silver stain. (g, h) Immunohistochemistry positive straining for IgG4 in plasma cells. IgG4: immunoglobulin G4. |
This is most consistent with IgG4-related TIN. IgG subclasses were checked and IgG4 was grossly elevated at 9.58 g/L (0.08 - 1.40). Serum lipase was normal. Abdominal CT showed no evidence of pancreatitis, lymphadenopathy or retroperitoneal fibrosis.
Treatment and outcome
He was commenced on prednisone 40 mg in November 2017. Creatinine level improved from over 500 µmol/L (eGFR 11 mL/min/1.73m2) to 346 µmol/L (eGFR 15 mL/min/1.73m2) in 6 weeks and continued to improve until it plateaued at about 260 µmol/L (eGFR 22 mL/min/1.73m2) (Fig. 2). Urine protein/creatinine ratio improved from 92 g/mol to 46 g/mol, and IgG4 dropped from 9.58 g/L to 2.47 g/L (Fig. 3).
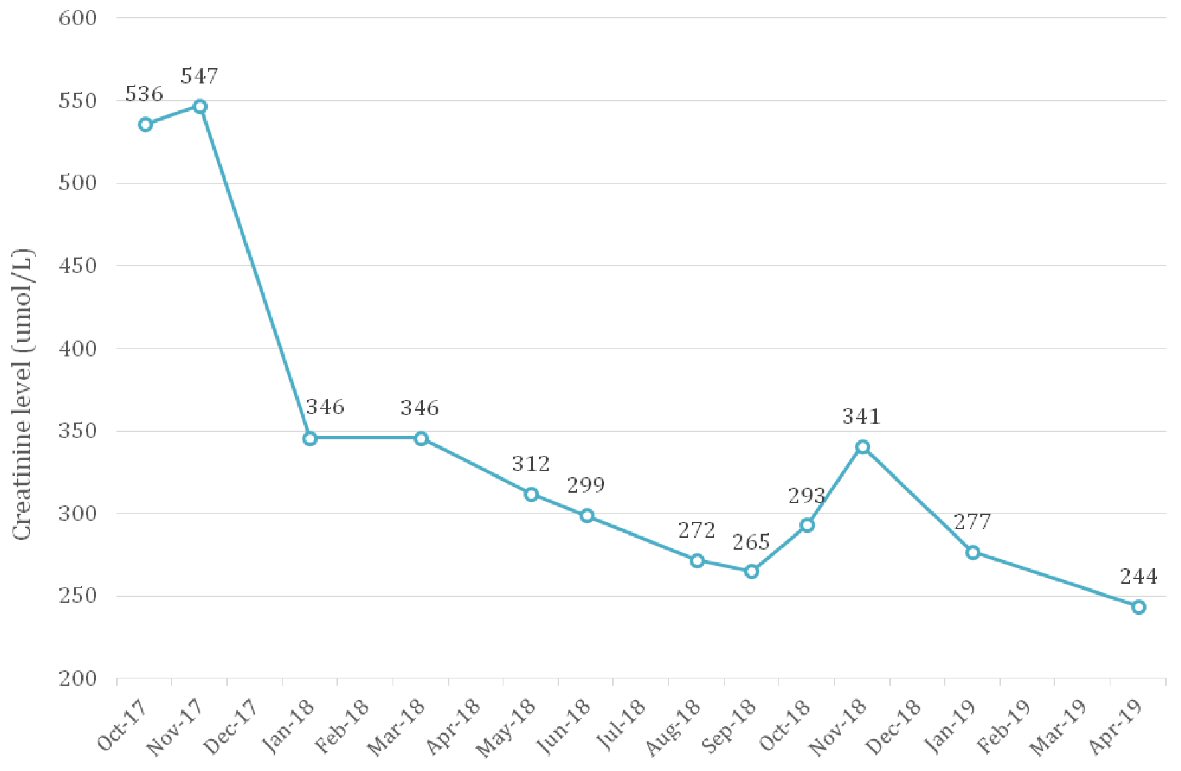 Click for large image | Figure 2. Creatinine trend from presentation in October 2017 to April 2019. Prednisone was commenced in November 2017 which coincided with the improvement in renal function seen then. |
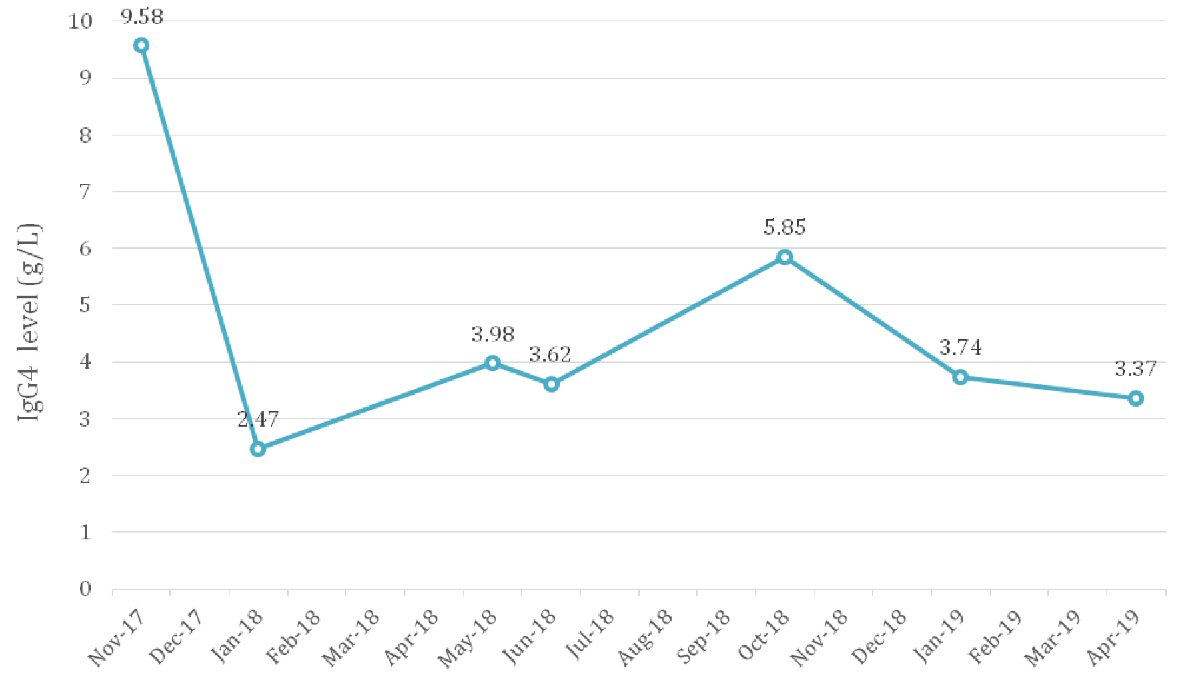 Click for large image | Figure 3. IgG4 levels from presentation in October 2017 to January 2019. IgG4: immunoglobulin G4. |
Prednisone was weaned down in April 2018 and stopped in June 2018. This was shortly followed by an isolated rise in IgG4 level up to 3 g/L without renal function deterioration. The patient was not keen on further treatment with corticosteroids due to marked adverse effects he had experienced earlier. Low-dose treatment with prednisone (5 mg) was reinstituted and azathioprine was commenced as a steroid-sparing agent in September 2018. However, the patient experienced unpleasant side effects of headache, myalgias and lethargy. Azathioprine was replaced with mycophenolate in October 2018. IgG4 level dropped from 5.85 g/L to 3.7 g/L. The patient remained in remission as of May 2019. He did not develop any other systemic manifestations of IgG4-RD.
| Discussion | ▴Top |
This case illustrates renal disease as a presenting and sole manifestation of IgG4-RD. Supportive evidence of this diagnosis in this case was the high serum IgG4, the histologic clues of severe intense Interstitial inflammation compared to typical drug-related interstitial nephritis, the IgG4-positive plasma cells infiltration in kidney, and Mott cells. No one of these findings alone is specific to IgG4-RD as it can be seen in autoimmune and allergic conditions; however the patient did not exhibit clinical, serological or historical evidence of autoimmune conditions or allergy.
IgG4-RD is a systemic inflammatory disorder which presents as an inflammatory mass-forming lesions, or organ enlargement. Unrecognised as a unified disease for well over a century, IgG4-RD has been likened to a “black crow flying through the dark night”. Thanks to better understanding of its pathologic basis and clinical presentations, many different disease entities merged into this condition (Table 1) [1].
 Click to view | Table 1. Conditions Once Regarded as Individual Disorders That Merged Into IgG4-Related Disease |
Patients often have multiple organ involvement simultaneously and sometimes metachronously, and spontaneous regression has been observed [1]. IgG4 disease has been described in nearly every organ system. Typical presentations include pancreatitis, lymphadenopathy, nephritis, retroperitoneal fibrosis, sialadenitis and sclerosing cholangitis [2-4]. As such, it can mimic many conditions including malignancy (e.g. lymphoma, respective solid organs), autoimmune conditions (e.g. Sjogren’s syndrome, sarcoidosis) and infections (e.g. tuberculosis, cytomegalovirus, Epstein-Barr virus, HIV). It often causes organ enlargement due to expanding inflammatory lesions, with solid organs like pancreas, kidneys and salivary glands becoming diffusely enlarged. Ductal organs, such as bile duct, bronchi, develop diffuse wall thickening leading to sclerosing cholangitis and bronchial thickening, respectively.
IgG4-related kidney disease (IgG4-RKD) manifestations include IgG4-TIN and membranous nephropathy [5]. The latter is associated with a negative M-type phospholipase A2 receptor (PLA2R), since PLA2R, an IgG4 antibody itself, is associated with primary membranous nephropathy. Renal insufficiency can also develop secondary to hydronephrosis associated with IgG4-related retroperitoneal fibrosis or lymphadenopathy [6]. Approximately 83% of patients have other organ system involvement [7], however, renal disease was the only manifestation in our patient.
Patients with IgG4-RKD present at an average age of 65 years, and 73-87% of them are men, and have high levels of serum IgG and IgG4, which is very similar to our case [7, 8]. The majority of the patients present with acute or progressive renal failure [7, 8]. Our patient had surprisingly low pyuria despite the significant TIN. IgG4-TIN is usually not accompanied by urinary excretion of many white blood cells (WBCs) or WBC casts. This finding probably reflects a mild tubulitis, a histopathological feature of IgG4-TIN, in contrast to drug-induced acute TIN [5]. There is a high frequency of hypocomplementemia associated with IgG4-TIN compared to other IgG4-RD manifestations [5, 7, 8], but is not found in all patients [7, 9]. This is interesting as IgG4 lacks potent complement activation property and may suggest that IgG4 is not pathological but rather an epiphenomenon. Complement levels often fall during disease activity, again consistent with our case, and hence that it can be a useful maker to monitor disease activity [10]. Likewise, serum immunoglobulin levels, especially IgG4, can be used to monitor disease activity [9]. Serum IgG4 levels usually rapidly decrease with corticosteroid therapy [7, 10].
Unlike our patient, 78% of patients usually have radiographic abnormalities characteristic of IgG4-RKD [7]. These typically include well defined circumscribed lesions seen on contrast-enhanced CT, or mass-like exophytic lesions protruding beyond the kidney surface. These discrete small nodules are within an otherwise unremarkable parenchyma, consistent with patchy site-selective involvement [5].
Typical biopsy findings are polyclonal lymphoplasmacytic infiltration, obliterative phlebitis, and storiform fibrosis. Some patchy fibrosis is present in all cases, which is generally more severe than that in other types of TIN, even in patients who present shortly after symptom onset, suggesting that the disease process starts much earlier than clinical manifestation. In our patient, the tubular atrophy and interstitial fibrosis might explain the partial renal function recovery, although the biopsy findings might not be representative of the whole renal parenchyma given the patchy disease involvement. The margin between affected and unaffected portions is very clearly demarcated, with this finding thought to correspond to the imaging feature of multiple low-density lesions [5]. When there is doubt whether plasma cell infiltration represents IgG4 disease or autoimmune conditions e.g. Sjogren’s syndrome, IgG4 immunohistochemistry seems to be a useful adjunctive test with a sensitivity of 100% and a specificity of 92% for IgG4-TIN [7]. This proved very useful in our case to delineate the etiology of the interstitial nephritis. An important differential here is ANCA-associated vasculitis, particularly eosinophilic granulomatosis with polyangiitis, which can sometimes show plasma cell infiltrates with IgG4 in the interstitium [11-13] with elevated serum IgG4 levels [14]. Clinical features, ANCA and C-reactive protein (CRP) levels (often not markedly raised in IgG4-RD) can help differentiate between the two conditions.
A good and rapid response to corticosteroid therapy is so commonly seen such that it is considered a criterion to support the diagnosis [5, 9, 15]. The extent of renal recovery depends on the degree of established and new fibrosis. Early rapid but partial recovery can be seen, especially if baseline eGFR is significantly reduced. This is consistent with histopathological localized severe fibrosis seen on repeat biopsy after long-term corticosteroid maintenance therapy, while other areas show only minor damage [16]. Likewise, on imaging, because of the patchy involvement, patchy recovery occurs. Contrast enhancement of the renal cortex recovers with therapy while other areas of the same kidney develop atrophic scarring with persistent decreased enhancement [16]. In our patient, the renal function plateaued at eGFR of 20 mL/min/1.73m2. Despite the significant established fibrosis, we continued immunosuppression not only to prevent renal relapse but also to prevent other IgG4-RD systemic manifestation.
Markers of response or relapse include renal function trend, serum total IgG and IgG4 and complement levels. If the disease is refractory or steroid dependent, addition of immunosuppressive agents is recommended [5]. B-cell depletion with rituximab has been used with success [17]. Reasonable second-line agents include azathioprine and mycophenolate as used in our patient. Recently, a randomized controlled trial demonstrated that combination corticosteroid and mycophenolate is superior to corticosteroid monotherapy and achieves less relapse rates [18]. More literature will become available in the future as our awareness and understanding of the condition improves.
Conclusions
IgG4-RD is an increasingly recognized systemic inflammatory disorder which can affect many organs. It can present in various non-specific ways leading to organ damage. Isolated renal involvement is rare, underrecognized and can lead to delay in diagnosis. Because early treatment with corticosteroids usually leads to prompt improvement, early diagnosis is important. Tissue biopsy is a key to diagnosis, and a high index of suspicion is required especially in absence of other systemic features.
Acknowledgments
Thanks to Dr Leo Francis, a senior pathologist at Anatomical Pathology, Pathology Queensland.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Written consent obtained from patient and is attached with the manuscript.
Author Contributions
M. Aljishi has drafted and written the manuscript and did the literature review and collected the images. He also took the patient consent. Z. Thet and T. Han provided recommendations and editions of manuscript. Z. Thet helped in patient’s follow-up and approached him for the consent. K. Madhan looked after the patient at presentation and made the diagnosis and management plan and followed him up. He also provided commentary on the manuscript.
| References | ▴Top |
- Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet. 2015;385(9976):1460-1471.
doi - Deshpande V, Chicano S, Finkelberg D, Selig MK, Mino-Kenudson M, Brugge WR, Colvin RB, et al. Autoimmune pancreatitis: a systemic immune complex mediated disease. Am J Surg Pathol. 2006;30(12):1537-1545.
doi pubmed - Uehara T, Hamano H, Kawa S, Sano K, Honda T, Ota H. Distinct clinicopathological entity 'autoimmune pancreatitis-associated sclerosing cholangitis'. Pathol Int. 2005;55(7):405-411.
doi pubmed - Kitagawa S, Zen Y, Harada K, Sasaki M, Sato Y, Minato H, Watanabe K, et al. Abundant IgG4-positive plasma cell infiltration characterizes chronic sclerosing sialadenitis (Kuttner's tumor). Am J Surg Pathol. 2005;29(6):783-791.
doi pubmed - Kawano M, Saeki T. IgG4-related kidney disease—an update. Curr Opin Nephrol Hypertens. 2015;24(2):193-201.
doi pubmed - Hamano H, Kawa S, Ochi Y, Unno H, Shiba N, Wajiki M, Nakazawa K, et al. Hydronephrosis associated with retroperitoneal fibrosis and sclerosing pancreatitis. Lancet. 2002;359(9315):1403-1404.
doi - Raissian Y, Nasr SH, Larsen CP, Colvin RB, Smyrk TC, Takahashi N, Bhalodia A, et al. Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol. 2011;22(7):1343-1352.
doi pubmed - Saeki T, Nishi S, Imai N, Ito T, Yamazaki H, Kawano M, Yamamoto M, et al. Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int. 2010;78(10):1016-1023.
doi pubmed - Kawano M, Saeki T, Nakashima H, Nishi S, Yamaguchi Y, Hisano S, Yamanaka N, et al. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol. 2011;15(5):615-626.
doi pubmed - Saeki T, Kawano M, Mizushima I, Yamamoto M, Wada Y, Nakashima H, Homma N, et al. The clinical course of patients with IgG4-related kidney disease. Kidney Int. 2013;84(4):826-833.
doi pubmed - Houghton DC, Troxell ML. An abundance of IgG4+ plasma cells is not specific for IgG4-related tubulointerstitial nephritis. Mod Pathol. 2011;24(11):1480-1487.
doi pubmed - Chang SY, Keogh KA, Lewis JE, Ryu JH, Cornell LD, Garrity JA, Yi ES. IgG4-positive plasma cells in granulomatosis with polyangiitis (Wegener's): a clinicopathologic and immunohistochemical study on 43 granulomatosis with polyangiitis and 20 control cases. Hum Pathol. 2013;44(11):2432-2437.
doi pubmed - Vaglio A, Strehl JD, Manger B, Maritati F, Alberici F, Beyer C, Rech J, et al. IgG4 immune response in Churg-Strauss syndrome. Ann Rheum Dis. 2012;71(3):390-393.
doi pubmed - Yamamoto M, Takahashi H, Suzuki C, Tabeya T, Ohara M, Naishiro Y, Yamamoto H, et al. Analysis of serum IgG subclasses in Churg-Strauss syndrome - the meaning of elevated serum levels of IgG4. Intern Med. 2010;49(14):1365-1370.
doi pubmed - Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366(6):539-551.
doi pubmed - Mizushima I, Yamada K, Fujii H, Inoue D, Umehara H, Yamagishi M, Yamaguchi Y, et al. Clinical and histological changes associated with corticosteroid therapy in IgG4-related tubulointerstitial nephritis. Mod Rheumatol. 2012;22(6):859-870.
doi pubmed - Khosroshahi A, Carruthers MN, Deshpande V, Unizony S, Bloch DB, Stone JH. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine (Baltimore). 2012;91(1):57-66.
doi pubmed - Yunyun F, Yu P, Panpan Z, Xia Z, Linyi P, Jiaxin Z, Li Z, et al. Efficacy and safety of low dose Mycophenolate mofetil treatment for immunoglobulin G4-related disease: a randomized clinical trial. Rheumatology (Oxford). 2019;58(1):52-60.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.