

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 11, Number 3, March 2020, pages 61-64
A Rare Case of Mainly Unilateral Focal Dermal Hypoplasia (Goltz Syndrome) in an Adult Male: A Case Report and Review of the Literature
Duncan Lyonsa, c, Christopher Rushtona, Sandeepal Sidhub
aGold Coast Hospital, Gold Coast, Queensland, Australia
bLaunceston General Hospital, Launceston, Tasmania, Australia
cCorresponding Author: Duncan Lyons, Gold Coast Hospital, Gold Coast, Queensland, Australia
Manuscript submitted February 20, 2020, accepted March 5, 2020
Short title: FDH in an Adult Male
doi: https://doi.org/10.14740/jmc3442
| Abstract | ▴Top |
Focal dermal hypoplasia (Goltz syndrome), is an exceedingly rare X-linked dominant genetic disorder. It is a multisystem disease, but it is hallmarked by characteristic skin changes. Focal dermal hypoplasia typically occurs in females (90%), and males are thought to only survive through having either a sporadic new mutation or somatic mosaicism. This report details a 48-year-old male diagnosed with predominately unilateral focal dermal hypoplasia that was reviewed decades post his initial diagnosis. He presented with multiple atrophic hyperpigmented macules and fat herniation along the lines of Blaschko, across primarily the right side of the body. Skin biopsy is the mainstay for the diagnosis and therefore dermatologists need to be aware of the classical cutaneous findings of familial dermal hypoplasia to ensure accurate diagnosis. Familial dermal hypoplasia is best managed through the collective minds of multidisciplinary teams.
Keywords: Focal dermal hypoplasia; Goltz syndrome; Male; Unilateral; X-linked dominant condition; Fat herniation
| Introduction | ▴Top |
Focal dermal hypoplasia (FDH), also known as Goltz syndrome, is a rare genetic disorder [1]. It is a multisystem disorder, classically affecting the skin, but also the teeth, eyes, face, skeletal, cardiovascular, central nervous and gastrointestinal systems [2]. It is inherited as an X-linked dominant condition, and it is characterised by typical reticulate or linear atrophic macules, papillomas, fat hernias and associated with multiple dental, skeletal and ocular defects [2]. This report describes an exceedingly rare case of congenital FDH, predominately affecting the right side of the body.
| Case Report | ▴Top |
A 48-year-old male was day 1 post a laparoscopic cholecystectomy and repair of umbilical hernia, where he additionally had an outpatient dermatology clinic for a periodic review of his FDH. He was diagnosed with FDH in early childhood at age 5. He had initially presented with asymptomatic non-healing skin lesions that affected mainly his right hand side of his body. Additionally, he was found to have syndactyly of hands and feet, digit hypoplasia, fingernail deformity, dental misalignment, hypodontia and nail thickening. He had normal milestones and development. His blood tests were normal, and a skin biopsy showed dermal atrophy with thin collagen fibres and impacting adipose tissue, which is consistent with a diagnosis of FDH. No molecular testing was completed.
Upon review during his appointment, he was noticed to have multiple atrophied hyper and hypopigmented macules, papules and fat herniation along the lines of Blaschko (Fig. 1a, b), predominately affecting the right side of the patient. Additional cutaneous examination revealed scattered telangiectasia (Fig. 2).
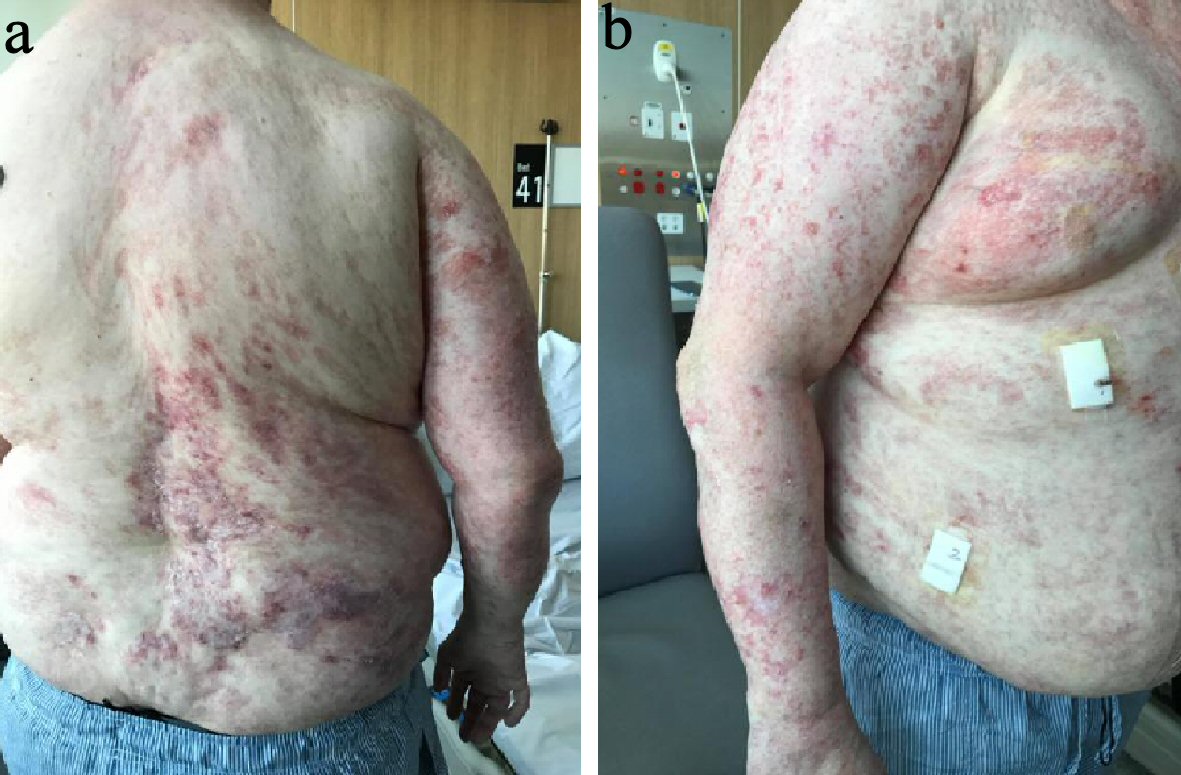 Click for large image | Figure 1. (a) Clinical photograph of the patient’s back. This photograph demonstrates the atrophy, pigmentation skin changes and fat herniation along the lines of Blaschko that are seen in FDH. These changes are seen predominately unilaterally, on the right side. (b) Clinical photograph of the patient’s right arm and torso. This photograph demonstrates the atrophy, pigmentation skin changes and fat herniation along the lines of Blaschko that are seen in FDH. FDH: focal dermal hypoplasia. |
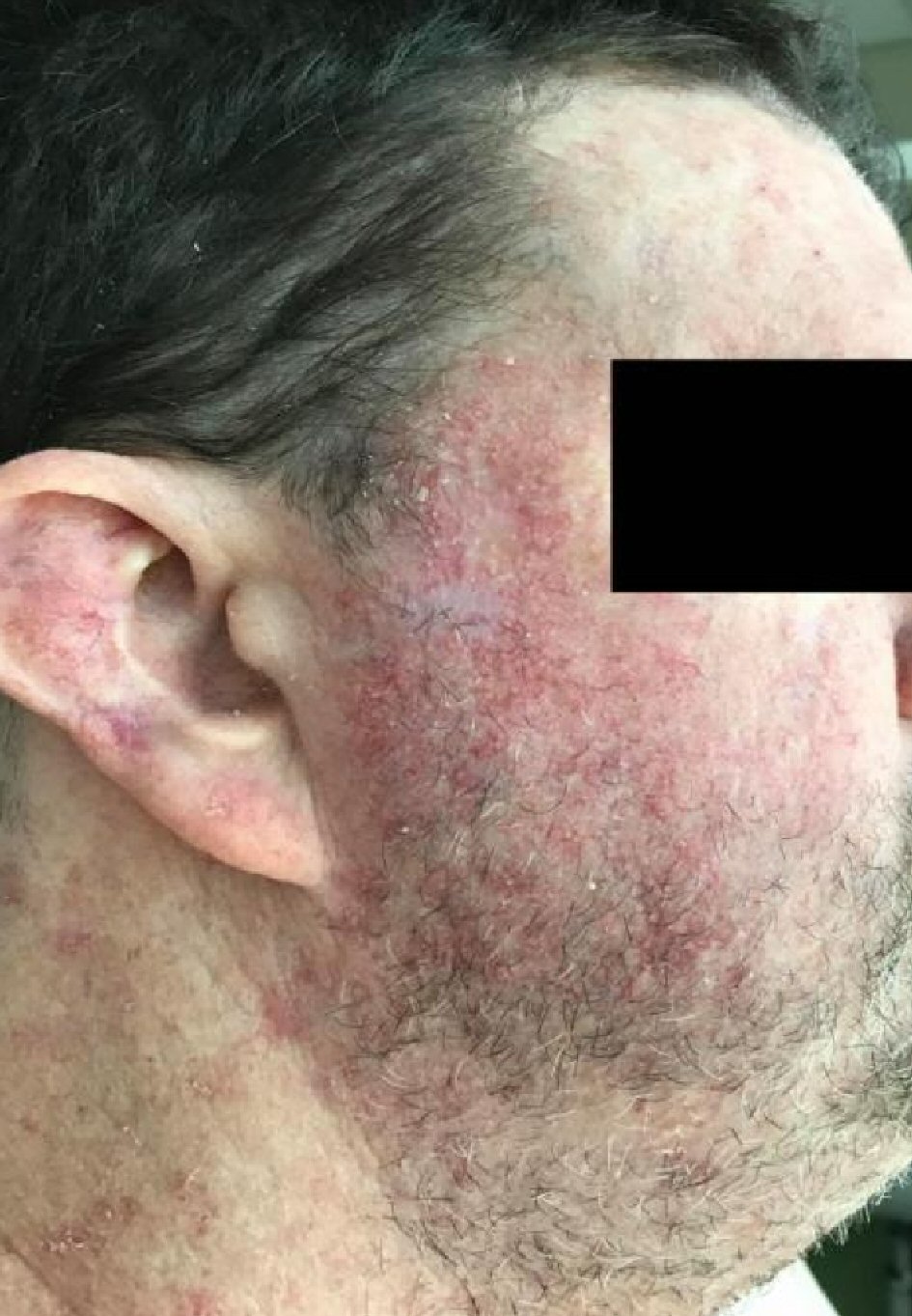 Click for large image | Figure 2. Clinical photograph of the patient’s right side of his face. The photograph demonstrates the telangiectasia that is seen in FDH across the cheek. FDH: focal dermal hypoplasia. |
He has a past history that includes gastroesophageal reflux disease, urinary tract infections, coronary artery bypass, basal cell carcinoma and squamous cell carcinoma. He has had multiple surgeries including an appendicectomy, tonsillectomy, repair of umbilical hernia, correction of dental alignment and surgical correction of finger and toe syndactyly. He currently takes esomeprazole, perindopril and panadeine forte and has given up on using regular moisturiser, as nothing in particular seems to aid his skin. He lives independently, works in administration and socially drinks alcohol and is a non-smoker. He is the first person in his family to be diagnosed with FSH, and he is yet to have children.
Mainly his general practitioner is currently managing him, with periodic input by a dermatologist. Ophthalmologists, pediatricians, oral and maxillary surgeons and general surgeons have all been involved in a multidisciplinary management approach.
| Discussion | ▴Top |
Although remarkably rare, FDH is a significant disorder that affects multiple body systems. It is an X-linked dominant condition, caused by mutations to the PORCN gene on chromosome Xp11.23. This is responsible for the encoding of a protein involved in Wnt signalling embryogenesis [3, 4]. The protein is responsible for the endoderm, mesoderm and ectoderm development during embryogenesis [3, 4]. FDH is normally lethal in males, and this is evidenced by still births, miscarriages and through the lack of transmission from father to son [5]. Only 10% of cases occur in males, and survival is thought to be due to either sporadic new mutation or somatic mosaicism [5-7]. Consequently, there are not many published case reports on male patients. The variations in the severity of FDH are thought to be caused by either post-zygotic genomic mosaicism or X chromosome lyonization [8].
FDH was first described by Goltz in 1962; he described three females who had severe congenital thinning of the skin and yellow papules, which represented herniation of adipose tissue [9]. The primary diagnostic features of FDH are skin changes [10]. These include patchy congenital skin aplasia following the lines of Blaschko (95%), which are white or pink in color and represent hypoplastic or atrophic areas of skin [11]. Congenital hyper- or hypopigmentation occurs in approximately 95% of patients, and again follows the lines of Blaschko. Fat nodules in the dermis of the trunk and extremities represent congenital fat herniation (65%). Other skin conditions include telangiectasias (80%), pebbled textured skin (60%), papillomas (60%) and photosensitivity [11]. Other cutaneous manifestations can include hair abnormalities including abnormalities of the hair shaft (85%), scalp alopecia (80%) and nail abnormalities that can include hypoplastic, dysplastic and ridged nails (85%) [10, 11].
The next most common extracutaneous manifestations are skeletal abnormalities; these are seen in approximately 50-90% of cases [5, 8, 11]. These include ectrodactyly, syndactyly, polydactyly, oligodactyly, digit hypoplasia and vertebral abnormalities. Additionally, dental manifestations can include dental crowding, hypodontia, supernumerary teeth, oligodontia, microdontia, enamel defects and root morphology abnormalities [12, 13]. Ocular manifestations can occur in FDH, ranging from 20% to 60% and can include microphthalmia, iris and chorioretinal colobomas, nystagmus, cataracts and strabismus [5, 8, 11]. Other abnormalities can include horseshoe kidneys, hernias, hearing defects, intellectual disability and congenital heart disease [11, 14]. From published research relating to FDH, there are only nine cases of unilateral/almost unilateral FDH, out of approximately 250 cases [15-22]. This report demonstrates a rare case of FDH presenting with unilateral symptoms and will subsequently only be the 10th unilateral FDH published in the literature and only the second male case.
The differential diagnosis of FDH includes incontinentia pigmenti, MIDAS (microphthalmia, dermal aplasia, and sclerocornea), Rothmund-Thomson syndrome, nevus lipomatosus superficialis, Adams-Oliver syndrome and aplasia cutis [23]. The gold standard for diagnosis includes a skin biopsy of an atrophic lesion [23]. The histopathology shows a thinned epidermis, hypoplastic dermis and the epidermis being impinged by adipose tissue [23, 24]. Scattered collagen bundles are seen on electron microscopy, and detection of the PORCN gene mutation through gene sequence analysis can also be performed [24]. Treatment of FDH involves mainly supportive therapy as currently; there is no effective treatment [25]. A multidisciplinary approach is imperative, and requires specialist teams including a dermatologist, pediatrician, ophthalmologist, plastic and orthopaedic surgeons.
Telangiectasia and papillomas can be treated with dye laser and cryotherapy [26]. The physical and functional problems related to FDH may require psychological input. Genetic counselling is imperative for preventative measures [3, 26]. For females, the prognosis is excellent and the life expectancy may be normal [3, 26]. The prognosis for males is utero lethality or if a mutation is present, 10% may survive. However, there is insufficient data pertaining to prognostic outcomes.
Conclusions
Although very rare, early recognition of familial dermal hypoplasia is important to ensure reduced morbidity and timely effective intervention. Skin biopsy is the mainstay for the diagnosis and therefore dermatologists need to be aware of the classical cutaneous findings of familial dermal hypoplasia to ensure accurate diagnosis. Familial dermal hypoplasia is best managed through the joint minds of multidisciplinary teams, which may include dermatologists, pediatricians, plastic and orthopaedic surgeons. Particularly in females, the prognosis is often good and the life expectancy may not be altered. Once diagnosed, genetic counselling is important for preventative measures.
Acknowledgments
None to declare.
Financial Disclosure
No sources of funding were utilized for the production of this manuscript.
Conflict of Interest
The authors declare that there is no conflict of interest regarding the publication of this article.
Informed Consent
Informed consent was gathered from the patient for publication of the case and accompanying photos.
Author Contributions
Duncan Lyons retrieved the case information and wrote the initial manuscript. Christopher Rushton and Sandeepal Sidhu edited the manuscript for submission.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Peters T, Perrier R, Haber RM. Focal dermal hypoplasia: report of a case with myelomeningocele, Arnold-Chiari malformation and hydrocephalus with a review of neurologic manifestations of Goltz syndrome. Pediatr Dermatol. 2014;31(2):220-224.
doi pubmed - Wright JT, Puranik CP, Farrington F. Oral phenotype and variation in focal dermal hypoplasia. Am J Med Genet C Semin Med Genet. 2016;172C(1):52-58.
doi pubmed - Alsharif S, Hindi S, Khoja F. Unilateral focal dermal hypoplasia (Goltz Syndrome): case report and literature review. Case Rep Dermatol. 2018;10(2):101-109.
doi pubmed - Grzeschik KH, Bornholdt D, Oeffner F, Konig A, del Carmen Boente M, Enders H, Fritz B, et al. Deficiency of PORCN, a regulator of Wnt signaling, is associated with focal dermal hypoplasia. Nat Genet. 2007;39(7):833-835.
doi pubmed - Goltz RW. Focal dermal hypoplasia syndrome. An update. Arch Dermatol. 1992;128(8):1108-1111.
doi pubmed - Paller AS. Wnt signaling in focal dermal hypoplasia. Nat Genet. 2007;39(7):820-821.
doi pubmed - Lasocki AL, Stark Z, Orchard D. A case of mosaic Goltz syndrome (focal dermal hypoplasia) in a male patient. Australas J Dermatol. 2011;52(1):48-51.
doi pubmed - Temple IK, MacDowall P, Baraitser M, Atherton DJ. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27(3):180-187.
doi pubmed - Goltz RW, Peterson WC, Gorlin RJ, Ravits HG. Focal dermal hypoplasia. Arch Dermatol. 1962;86:708-717.
doi pubmed - Ghoshal B, Lahiri S, Nandi D. A case of male goltz syndrome. Case Rep Pediatr. 2012;2012:728509.
doi pubmed - Bostwick B, Van den Veyver I, Sutton V. Seattle: focal dermal hypoplasia. GeneReviews. University of Washington. 1993.
- Murakami C, de Oliveira Lira Ortega A, Guimaraes AS, Goncalves-Bittar D, Bonecker M, Ciamponi AL. Focal dermal hypoplasia: a case report and literature review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;112(2):e11-18.
doi pubmed - Tejani Z, Batra P, Mason C, Atherton D. Focal dermal hypoplasia: oral and dental findings. J Clin Pediatr Dent. 2005;30(1):67-72.
doi pubmed - Riyaz N, Riyaz A, Chandran R, Rakesh SV. Focal dermal hypoplasia (Goltz syndrome). Indian J Dermatol Venereol Leprol. 2005;71(4):279-281.
doi pubmed - Lee S, Choe SJ, Ahn SK. Almost unilateral focal dermal hypoplasia. Ann Dermatol. 2017;29(1):91-94.
doi pubmed - Stalder JF, Delaire J, David A, Cohen JY, Le Pape A. [Unilateral Goltz syndrome in a boy]. Ann Dermatol Venereol. 1984;111(9):829-830.
- Denis-Thely L, Cordier MP, Cambazard F, Misery L. [Unilateral focal dermal hypoplasia]. Ann Dermatol Venereol. 2002;129(10 Pt 1):1161-1163.
- Aoyama M, Sawada H, Shintani Y, Isomura I, Morita A. Case of unilateral focal dermal hypoplasia (Goltz syndrome). J Dermatol. 2008;35(1):33-35.
doi pubmed - Fernandez-Torres R, Del Pozo J, Garcia-Silva J, Fonseca E. [Unilateral focal dermal hypoplasia]. Actas Dermosifiliogr. 2010;101(1):96-98.
doi pubmed - Tenkir A, Teshome S. Goltz syndrome (focal dermal hypoplasia) with unilateral ocular, cutaneous and skeletal features: case report. BMC Ophthalmol. 2010;10:28.
doi pubmed - Maalouf D, Megarbane H, Chouery E, Nasr J, Badens C, Lacoste C, Grzeschik KH, et al. A novel mutation in the PORCN gene underlying a case of almost unilateral focal dermal hypoplasia. Arch Dermatol. 2012;148(1):85-88.
doi pubmed - Nakanishi G, Hasegawa K, Oono T, Koshida S, Fujimoto N, Iwatsuki K, Tanaka H, et al. Novel and recurrent PORCN gene mutations in almost unilateral and typical focal dermal hypoplasia patients. Eur J Dermatol. 2013;23(1):64-67.
doi pubmed - Jain A, Chander R, Garg T, Nikita, Shetty GS. A rare multisystem disorder: Goltz syndrome - case report and brief overview. Dermatol Online J. 2010;16(6):2.
- del Carmen Boente M, Asial RA, Winik BC. Focal dermal hypoplasia: ultrastructural abnormalities of the connective tissue. J Cutan Pathol. 2007;34(2):181-187.
doi pubmed - Ghosh SK, Dutta A, Sarkar S, Nag SS, Biswas SK, Mandal P. Focal dermal hypoplasia (Goltz Syndrome): a cross-sectional study from Eastern India. Indian J Dermatol. 2017;62(5):498-504.
doi pubmed - Bharani S, Thakkar S. A case report of focal dermal hypoplasia-Goltz syndrome. Indian Dermatol Online J. 2013;4(3):241-243.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.