

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 4, Number 1, January 2013, pages 52-55
Primary Hepatic Lymphoma: A Diagnosis to Remember
Ben Warnera, b, George Birda, John Schofielda, Saad Rassama
aMaidstone Hospital, Hermitage Lane, Maidstone, ME16 9QQ, UK
bCorresponding author: Ben Warner, Maidstone Hospital, Hermitage Lane, Maidstone, ME16 9QQ, UK
Manuscript accepted for publication October 4, 2011
Short title: Primary Hepatic Lymphoma
doi: https://doi.org/10.4021/jmc345w
| Abstract | ▴Top |
We present 2 cases of PHL presenting to our hospital in the last 6 months. Both cases had similarities in that they had a rapidly progressive time course of less than 8 weeks and radiology failed to illustrate the normal disease pattern. Both were elderly men presenting with jaundice, rare presentations of PHL. One patient was interesting in that he had a bicytopenic blood picture, a common manifestation of PHL, whilst his lactate dehydrogenase was normal. The other had a past history of hepatitis B, interesting for its possible association with PHL. Whilst one patient died prior to treatment, the other was successfully treated with aggressive multidrug chemotherapy. A liver biopsy is essential in any patient where there is a high index of suspicion for PHL based on a bicytopenic blood picture, abnormal liver function tests or radiology. If diagnosed early enough, chemotherapy has proven a successful treatment.
Keywords: Primary hepatic lymphoma; PHL; Hepatitis B
| Introduction | ▴Top |
Primary Hepatic Lymphoma (PHL) is a rare type of extra nodal lymphoma accounting for less than one percent of all lymphomas [1]. It presents with a wide range of subtle manifestations and for this reason may be late or missed altogether. Diagnosis is confirmed only by liver biopsy and a review of the literature shows success with certain surgical and chemotherapeutic treatments. Late diagnosis, however, or failure to make the diagnosis can end in fulminant hepatic failure and ultimately death. We present 2 cases of PHL admitted to our district general hospital in the last 6 months and discuss the aetiology, presentations, investigations and treatments of this rare disease.
| Case Report | ▴Top |
Case 1
A 73 years old male presented with a 10 day history of jaundice, pale stools, dark urine and diarrhoea with some pain in the right upper quadrant. His past medical history included osteoarthritis, hypertension and hepatitis B from which he had recovered (HBsAg negative). His medications included lisinopril, spironolactone, omeprazole and ibuprofen. He denied drinking any alcohol or smoking. On examination, he was markedly jaundiced with tenderness in the right upper quadrant but had no organomegaly. Bloods tests on admission showed a bilirubin of 122 µmol/L (NR 2 - 17), alkaline phosphatase (ALP) 346 U/L (NR 35 - 104), aspartate transaminase (AST) 104 U/L (4 - 31), albumin 40 g/L (34 - 48), INR 1.1 (0.7 - 1.2), haemoglobin (Hb) 12.6 g/dL (11.5 - 16.5), white blood cell (WBC) 8.6 K/µL (3.4 - 11), platelets (Plts) 358 K/µL (140 - 450), Calcium (Ca) 3.52 mmol/L (2.3 - 2.75). An ultrasound of his liver revealed a course echotexture suggestive of cirrhosis with moderate ascites but no duct dilatation. Serology for Hepatitis B sAg, Hepatitis C, and Hepatitis A were negative and levels of Alphafetoprotein (AFP), Caeruloplasmin, and Copper were normal. Liver biopsy confirmed the patient to have Non-Hodgkins Lymphoma staining positive for CD20 lymphocytes (Fig. 1-4). He was commenced on a combination chemotherapy of Rituximab, Chlorambucil, Doxorubicin, Vincristine and Prednisolone (RCHOP) and went into tumor lysis syndrome with acute renal failure requiring admission to intensive care. However, he made a good recovery and within 4 months his liver function had normalized.
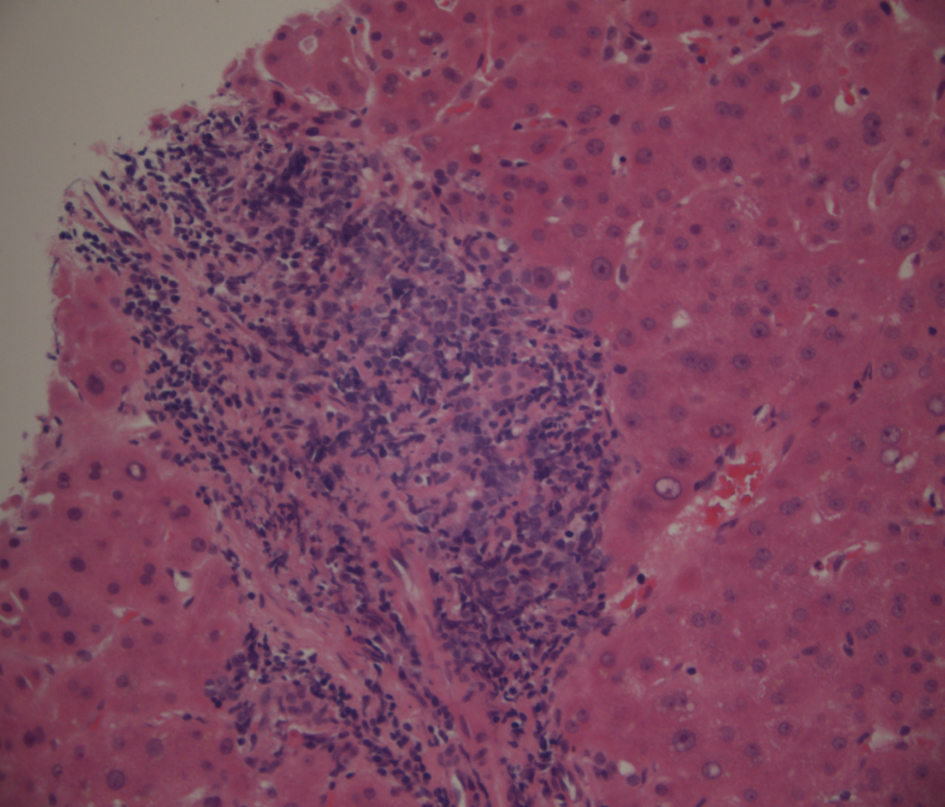 Click for large image | Figure 1. Low power view of portal tract lymphoma stained with haematoxylin and eosin. |
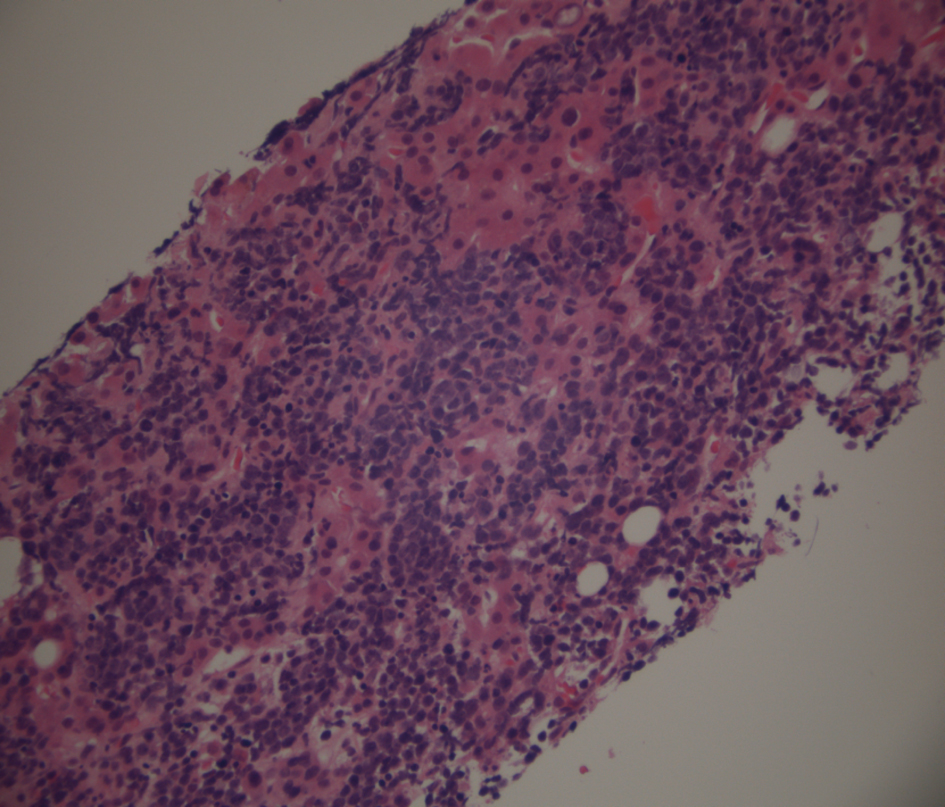 Click for large image | Figure 2. Low power view of liver parenchymal lymphoma stained with haematoxylin and eosin. |
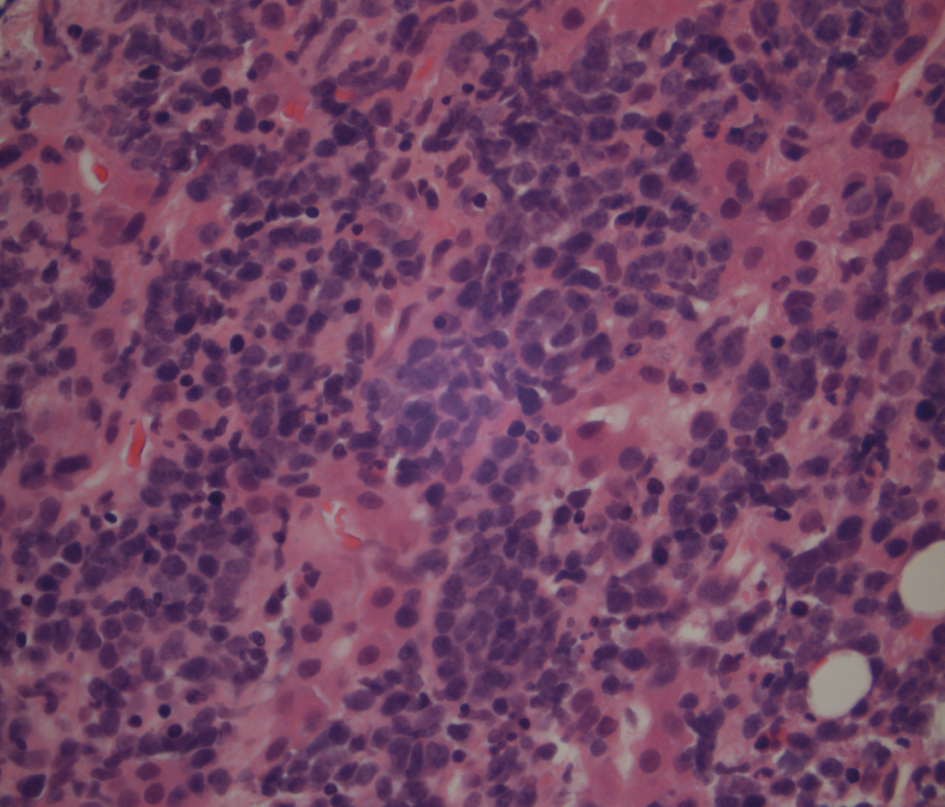 Click for large image | Figure 3. Liver biopsy showing a higher power of the lymphoma cells which have hyperchromatic nuclei and little cytoplasm. |
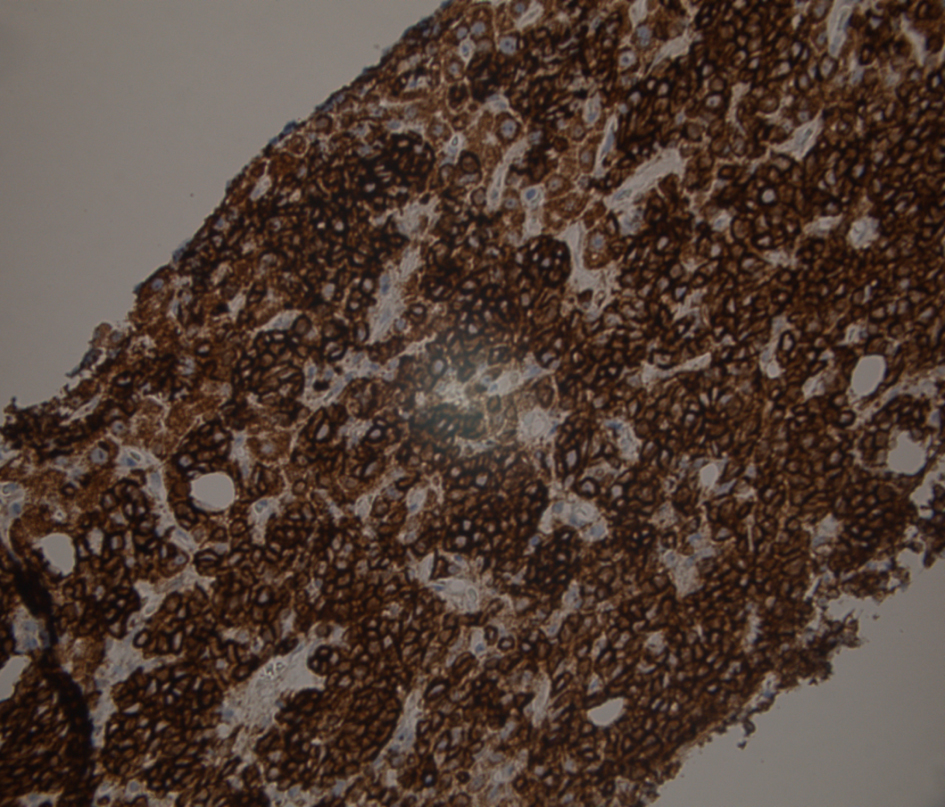 Click for large image | Figure 4. Liver biopsy immunostained for CD20, the usual B lymphocyte marker. Shows strong membrane and cytoplasmic positivity confirms that this is a B cell lymphoid proliferation. |
Case 2
An 80 years old male presented with a month’s history of diarrhoea 5 times daily with loose offensive stool. He had a week history of jaundice and was admitted urgently by his General Practitioner for investigation. His past medical history included chronic obstructive airways disease for which he took inhalers. He admitted to consuming 10 units of alcohol per week and was a non-smoker. On examination, he was jaundiced, cachectic with moderate ascites but with no organomegally. Blood tests showed a bilirubin of 28 µmol/L, ALP 187 U/L, AST 113 U/L, alb 24 g/L, INR 1.6, Hb 12.4 g/dL, plts 91 K/µL and WBC 2.26 K/µL. A sigmoidoscopy showed an early diverticular stricture preventing further examination for the cause of his diarrhoea. A CT abdomen showed an enlarged liver edge, moderate ascites, 16 cm splenomegaly and some mildly enlarged nodes around the coeliac axis and porta hepatis measuring 15 mm. Autoimmune screen, hepatitis serology screen were all negative and Caeruloplasmin, AFP, Immunoglobulins and Carcinoembryonic Antigen (CEA) were all normal. Ferritin was 758 ng/mL. Investigation of this patient’s bicytopenia showed a normal blood film, reticulocyte count of 36 and a lactate dehydrogenase (LDH) of 348 U/L. He underwent paracentesis, the fluid protein content being 6 g/L, followed by a liver biopsy. He was discharged on a salt restricted diet, diuretics and dietary supplements with a view to follow-up. Unfortunately, he was readmitted with sepsis and uncontrolled fast AF with respiratory compromise and died 2 weeks later. Liver biopsy result showed him to have a low grade marginal zone lymphoma staining positive for CD20+ lymphocytes.
| Discussion | ▴Top |
As stated already PHL is rare. PHL is different from other lymphomas which involve the Liver in 50% of cases [2] in that in order to make a diagnosis they must demonstrate no nodal involvement, involve predominantly the liver and must be in the absence of any evidence of leukaemia. Both of our cases meet this definition.
Since clinical features and laboratory tests are wide ranging liver biopsy is essential in confirming a suspected diagnosis.
Diffuse large B cell is the most likely pathology followed by Small cell lymphocytic, T cell (Fig. 5), follicular and marginal zone lymphoma in decreasing prevalence [1, 3]. The majority of PHL patients (67%) are middle-aged men (median age 50 years). Presentations vary significantly but hepatomegally (80% of cases), weight loss (47% of cases), and fevers (37-86%) are the most common. Jaundice is actually rare (4%) and symbolises diffuse hepatic involvement [4]. Fulminant hepatic failure is very rare at first presentation. Liver function tests tend to be cholestatic in the absence of duct dilatation on ultrasound. LDH is raised in 85% of cases [2]. A full blood count often demonstrates a pancytopenic or bicytopenic picture, pancytopenia occurring most likely in T cell lymphoma. AFP and CEA are both normal distinguishing this from the other differential diagnoses of HCC or metastatic Gastrointestinal cancer. A raised calcium is seen in 40% [4] of cases and is put down to the production of Calcitriol by malignant cells.
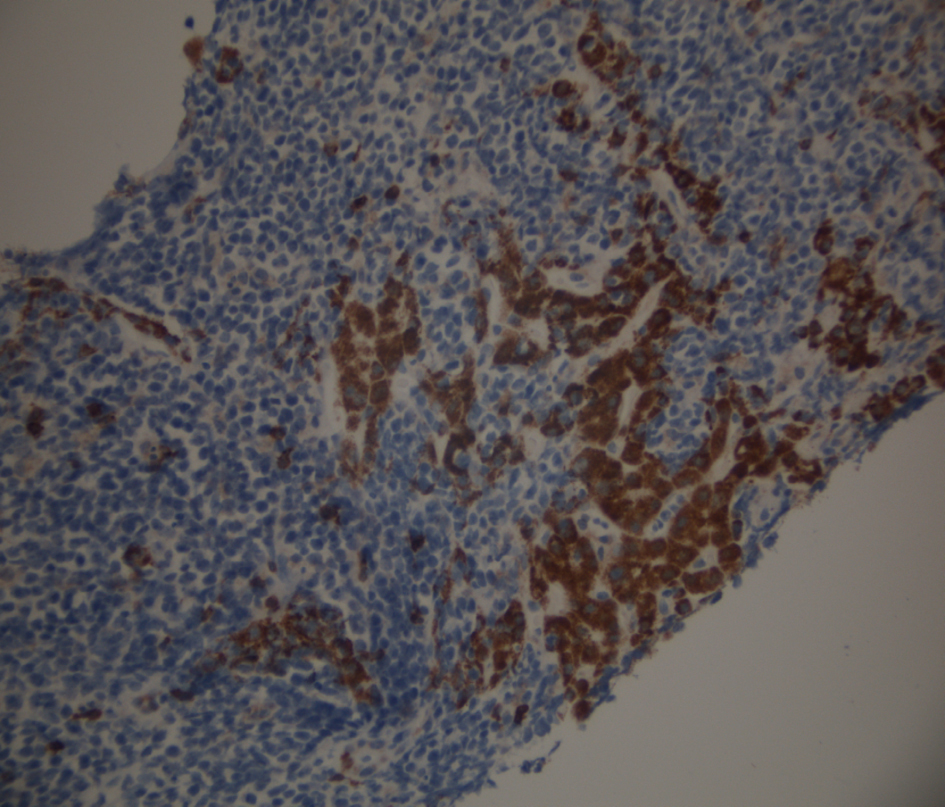 Click for large image | Figure 5. Liver biopsy stained for CD3, a T cell marker which is negative in the tumor cells. There is aberrant staining in hepatocytes due to incomplete blocking of endogenous peroxidases in the cells. |
Both Ultrasound and CT help to diagnose the disease with solitary tumors being seen in 42% of cases, multiple lesions in 50% and diffuse disease in 8% [4] though the latter is commoner amongst Chinese. The pattern of infiltration bears no prognostic value. Whereas Hepatocellular Carcinoma (HCC) appears isodense on CT with rim enhancement in the arterial phase, PHLs are hypodense and rarely enhance and this allows distinction between these two types of primary liver cancers [5, 6]. Liver biopsy remains the mainstay of diagnosis.
Treatment revolves around 3 modalities: Surgery, radiotherapy and chemotherapy. Although traditionally resection followed by RCHOP chemotherapy has been the mainstay of treatment multidrug regimens alone, such as alternating triple combinations (ATT), have shown more success in reaching remission. A study by Page et al [7] treated 24 patients with PHL by giving them aggressive combination chemotherapy regimens without surgery and had remission rates of 83%.
In fact, these cases share very little in common other than the presentation of jaundice and the speed at which their disease progressed. Both patients had extensive disease within 2 months of the first onset of symptoms. Neither patient had fulminant hepatic failure which is a hallmark of other liver pathologies. Radiology demonstrated no single or multiple lesions which would normally be expected. The diagnosis was only made following a biopsy.
Hepatitis B has been speculated as playing a role in giving rise to PHL [8] and it is interesting that one of our cases had hepatitis B in the past. Although he was HbsAg negative his viral load was never checked and therefore we can never be certain that he was without disease. Indeed, in the case from Matano et al [9] immunohistochemical staining demonstrated no replicating virus within the neoplastic cellls although they hypothesised that chronic antigenic stimulation by the virus could be associated with the development of lymphoma rather like other immunological conditions such as Hashimotos thyroiditis and Sjogrens syndrome [10] have been linked to the development of extra nodal lymphomas through the local proliferation of lymphoid tissues. Hepatitis B has a strong correlation with hepatocellular carcinoma though no studies have provided a concrete link with lymphoma. Cirrhosis itself has been linked to extra nodal lymphoma in a number of cases [11-13]. Heimann et al [11] describe 49 patients with lymphoma, 8% of whom had cirrhosis. Hepatitis C and EBV have both been associated with the development of monoclonal B-lymphoproliferative disorders [14, 15]. Mixed cryoglobulinaemia (MC) is seen by many as a variant of low grade B cell lymphoma [16] and antibodies to hepatitis C have been found in 98% of patients with type 2 MC. Bronowicki et al [17] explored further the relationship between PHL and hepatitis C and a retrospective study of 31 patients with PHL showed 6 (20%) were positive for the virus. Likewise Page et al [7] showed that 6 out of 10 of their 24 patients with PHL had hepatitis C.
Median survival has been reported as between 8 and 16 months with remission rates of less than 20% [1]. Poor prognostic factors include diffuse infiltration, high Ki index (Fig. 6), increased age, raised LDH, cirrhosis and presence of raised alpha-2 microglobulin.
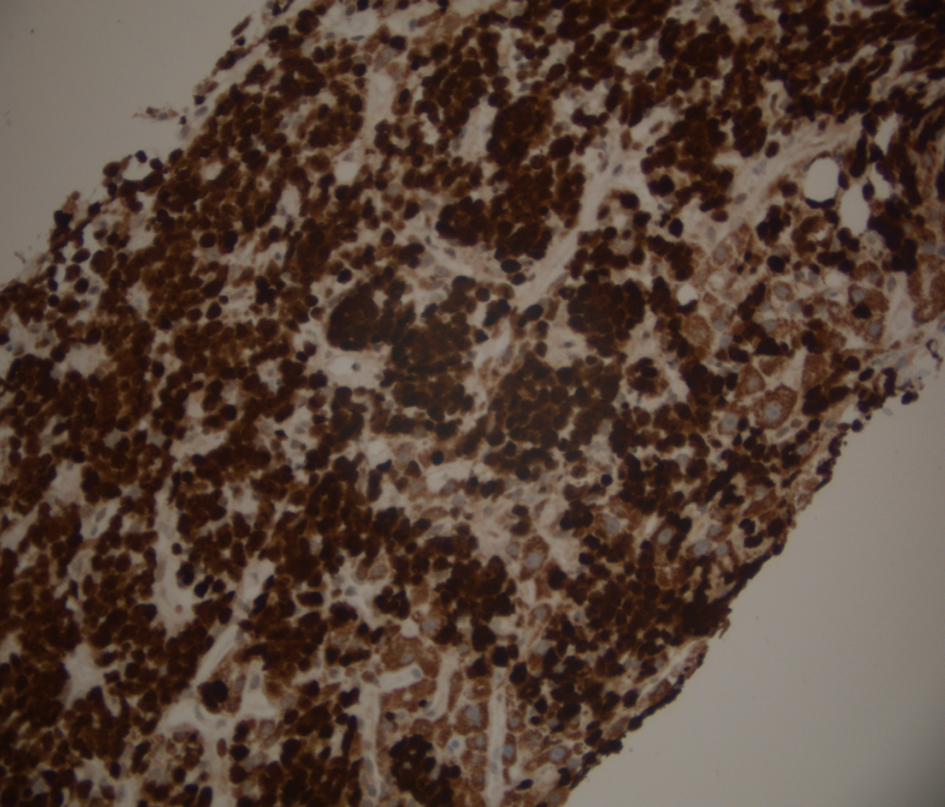 Click for large image | Figure 6. Liver biopsy stained for Mib-1 (Ki 67), a proliferation marker which is positive in the nuclei of cells in the mitotic cycle. In this case the proliferation index is nearly 100%, a finding associated with very aggressive lymphomas including Burkitts lymphoma and other diffuse large B cell lymphomas. |
To conclude with, there are few similarities between cases reported within the literature yet early detection of the disease yields successful results with standard chemotherapy regimes and therefore it is a disease for which we should have a high index of suspicion. Liver biopsy is suggested in any patient that presents with abnormal liver function tests, raised LDH and a bicytopenic/pancytopenic picture in the context of isolated hepatic disease radiologically. Hepatitis B and C may be an indirect aetiology through chronic antigen stimulation. The disease is aggressive with a short time course and RCHOP chemotherapy is the most suitable initial treatment although there is future hope with more aggressive regimes.
| References | ▴Top |
- Haider FS, Smith R, Khan S. Primary hepatic lymphoma presenting as fulminant hepatic failure with hyperferritinemia: a case report. J Med Case Rep. 2008;2:279.
pubmed - Delshad SD, Ahdoot JJ, Portocarrero DJ. Primary hepatic lymphoma. Clin Gastroenterol Hepatol. 2010;8(5):e49-50.
pubmed doi - Gomyo H, Kagami Y, Kato H, Kawase T, Ohshiro A, Oyama T, Kamiya Y, et al. Primary hepatic follicular lymphoma : a case report and discussion of chemotherapy and favorable outcomes. J Clin Exp Hematop. 2007;47(2):73-77.
pubmed doi - Agmon-Levin N, Berger I, Shtalrid M, Schlanger H, Sthoeger ZM. Primary hepatic lymphoma: a case report and review of the literature. Age Ageing. 2004;33(6):637-640.
pubmed doi - Nonami A, Takenaka K, Harada N, Kono K, Kamezaki K, Numata A, Karube K, et al. Primary hepatic lymphoma 1 year after resection of hepatocellular carcinoma. J Clin Oncol. 2006;24(36):5784-5786.
pubmed doi - Maher MM, McDermott SR, Fenlon HM, Conroy D, O'Keane JC, Carney DN, Stack JP. Imaging of primary non-Hodgkin's lymphoma of the liver. Clin Radiol. 2001;56(4):295-301.
pubmed doi - Page RD, Romaguera JE, Osborne B, Medeiros LJ, Rodriguez J, North L, Sanz-Rodriguez C, et al. Primary hepatic lymphoma: favorable outcome after combination chemotherapy. Cancer. 2001;92(8):2023-2029.
pubmed doi - Talamo TS, Dekker A, Gurecki J, Singh G. Primary hepatic malignant lymphoma: its occurrence in a patient with chronic active hepatitis, cirrhosis, and hepatocellular carcinoma associated with hepatitis B viral infection. Cancer. 1980;46(2):336-339.
pubmed doi - Matano S, Nakamura S, Annen Y, Hattori N, Kiyohara K, Kakuta K, Kyoda K, et al. Primary hepatic lymphoma in a patient with chronic hepatitis B. Am J Gastroenterol. 1998;93(11):2301-2302.
pubmed doi - Talal N, Sokoloff L, Barth WF. Extrasalivary lymphoid abnormalities in Sjogren's syndrome (reticulum cell sarcoma, "pseudolymphoma," macroglobulinemia). Am J Med. 1967;43(1):50-65.
pubmed doi - Heimann R. Cirrhosis and lymphoproliferative disorders. Lancet. 1971;2(7726):705.
pubmed doi - Naparstek J, Eliakim M. Malignant lymphoproliferative disorders in chronic liver disease. Report of four cases and review of the literature. Am J Dig Dis. 1978;23(10):887-892.
pubmed doi - Viteri A, Vernace SJ, Schaffner F. Extrahepatic malignancy in chronic liver disease: report of six cases. Gastroenterology. 1976;71(6):1075-1078.
pubmed - Pozzato G, Mazzaro C, Crovatto M, Modolo ML, Ceselli S, Mazzi G, Sulfaro S, et al. Low-grade malignant lymphoma, hepatitis C virus infection, and mixed cryoglobulinemia. Blood. 1994;84(9):3047-3053.
pubmed - Schweiger F, Shinder R, Rubin S. Primary lymphoma of the liver: a case report and review. Can J Gastroenterol. 2000;14(11):955-957.
pubmed - Pawlotsky JM, Roudot-Thoraval F, Simmonds P, Mellor J, Ben Yahia MB, Andre C, Voisin MC, et al. Extrahepatic immunologic manifestations in chronic hepatitis C and hepatitis C virus serotypes. Ann Intern Med. 1995;122(3):169-173.
pubmed - Bronowicki JP, Bineau C, Feugier P, Hermine O, Brousse N, Oberti F, Rousselet MC, et al. Primary lymphoma of the liver: clinical-pathological features and relationship with HCV infection in French patients. Hepatology. 2003;37(4):781-787.
pubmed doi
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.