

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 12, Number 3, March 2021, pages 107-111
Immunoglobulin G4 Related-Disease: A Rare Presentation With Secondary Hypereosinophilic Syndrome and Eosinophilic Ascites
Luisa Serpa Pintoa, f, Nuno Jorge Lamasb, c, d, Ana Campare, Alvaro Ferreiraa, e, Ana Rita Cruza
aInternal Medicine Department, Centro Hospitalar e Universitario do Porto, Porto, Portugal
bAnatomic Pathology Service, Pathology Department, Centro Hospitalar e Universitario do Porto, Porto, Portugal
cLife and Health Sciences Research Institute (ICVS), School of Medicine, University of Minho, Braga, Portugal
dICVS/3Bs PT Government Associate Laboratory, Braga/Guimaraes, Portugal
eClinical Immunology Unit, Centro Hospitalar e Universitario do Porto, Porto, Portugal
fCorresponding Author: Luisa Serpa Pinto, Internal Medicine Department, Centro Hospitalar e Universitario do Porto, Largo do Prof. Abel Salazar, 4099-001 Porto, Portugal
Manuscript submitted December 12, 2020, accepted December 19, 2020, published online January 19, 2021
Short title: IgG4-RD Presented With HES
doi: https://doi.org/10.14740/jmc3634
| Abstract | ▴Top |
Immunoglobulin G4 related-disease (IgG4-RD) is a multisystemic immune-mediated fibroinflammatory disease, with a strong predilection for salivary and lacrimal glands, pancreas, biliary tree, lungs, kidneys, aorta, and retroperitoneum. In the case of pancreatic involvement, it manifests as autoimmune pancreatitis. Patients with IgG4-RD usually have mild to moderate eosinophilia in the peripheral blood, however, they may present a secondary hypereosinophilic syndrome (HES). Although there are cases described with severe eosinophilia (> 5,000/µL), the 2019 American College of Rheumatology/European League Against Rheumatism classification criteria for IgG4-RD considers peripheral eosinophilia above 3,000/µL as an exclusion criterion, although stating that assessment for the presence of exclusion criteria should be individualized depending on a patient’s clinical scenario. Here, we describe the clinical case of a 30-year-old woman who presented with chronic diarrhea and abdominal pain. The diagnostic workup revealed eosinophilic ascites, severe peripheral blood eosinophilia (> 5,000/µL), IgG4 elevation (> 2 × upper normal limit), and also diffuse swelling in the body and pancreatic tail (computed tomography (CT) scan). There was a prompt response to corticosteroid therapy with clinical resolution and continued remission under therapy. The patient was diagnosed with IgG4-RD with secondary HES, explaining the gastrointestinal tract and peritoneum damage in the form of enterocolitis and ascites. Exclusion of alternative diagnosis was made.
Keywords: IgG4 related-disease; Hypereosinophilic syndrome; Severe eosinophilia; Autoimmune pancreatitis; Eosinophilic ascites
| Introduction | ▴Top |
Immunoglobulin G4 related-disease (IgG4-RD) is a multisystemic immune-mediated fibroinflammatory disease, with a broad spectrum of organ system involvement [1]. IgG4-RD is a relapsing and remitting condition clinically characterized by mass-forming lesions, which has a strong predilection for salivary and lacrimal glands, pancreas, biliary tree, lungs, kidneys, aorta, and retroperitoneum, increased serum IgG4 concentration, and prompt response to glucocorticoid treatment. The histopathological diagnosis of IgG4-RD rests on the combined presence of a dense lymphoplasmacytic infiltrate, a storiform pattern of fibrosis, and obliterative phlebitis together with increased numbers of IgG4+ plasma cells [2].
As autoimmune pancreatitis (AIP), pancreatic involvement was among the first described manifestations of the disease and is one of the most commonly involved organs [3]. Clinically, it may present with or without painless jaundice, with the development of hyperglycemia and steatorrhea being uncommon. The most frequently used diagnostic criteria for AIP, under the acronym HISORt, are based on the presence of characteristic histological and imaging features, serology, other organ involvement, and adequate response to corticosteroids [4].
Secondary peripheral blood eosinophilia is a common finding in patients with IgG4-RD [5], and patients previously classified as having idiopathic secondary hypereosinophilic syndrome (HES) have been reclassified as having IgG4-RD [6]. Eosinophilia is usually mild to moderate, with cases described with severe eosinophilia (absolute eosinophil count (AEC) > 5 × 103/µL) [5-7]. On the other hand, the 2019 American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) classification criteria for IgG4-RD consider peripheral eosinophilia above 3,000/µL as exclusion criteria depending on the clinical scenario [1].
We describe a case of a patient presenting IgG4-RD with secondary HES, with severe eosinophilia causing several organ damage types due to tissue infiltration of eosinophils, explaining the gastrointestinal tract and peritoneum damage in the form of enterocolitis and eosinophilic ascites.
| Case Report | ▴Top |
We present a case of a 30-year-old woman with a history of allergic rhinitis and atopy without current medication. She was admitted to our service with chronic diarrhea (started 2 months earlier), with more than seven mucus stools daily, abdominal pain, nausea, vomiting, and increased abdominal perimeter. The analytical study performed 1 month earlier following a primary care physician visit showed a hemoglobin of 12.5 g/dL (normal range (NR): 12 - 15 g/dL), white blood cells (WBC) 8.46 × 103/µL (NR: 4 - 11.0 × 103/µL), AEC 2.02 × 103/µL (NR: 0.04 - 0.40 × 103/µL) (24%, NR: 1.0-6.0%), platelets 260 × 103/µL (NR: 150 - 400 ×103/µL). Besides, the abdominal ultrasound revealed small volume ascites. Parasites were not found in the three stool samples analyzed. She had already undergone several treatments for infectious gastroenteritis without any improvement, including treatment with ciprofloxacin and albendazole as an outpatient. The patient had performed an abdominal-pelvic computed tomography (CT) scan, which revealed high-volume ascites, pronounced intestinal wall parietal thickening in the small intestine, and diffuse mesenteric densification. She was referred to the emergency department (ED) on the same day.
Upon admission to the ED, she presented a blood pressure of 102/62 mm Hg, a heart rate of 88 bpm, and abdominal distension compatible with high-volume ascites. The patient did not have fever. Reviewing the analytical study performed we obtained: WBC 12.32 × 103/µL (NR: 4 - 11.0 × 103/µL), AEC 2.06 × 103/µL (NR: 0.04 - 0.40 × 103/µL), hemoglobin 13.9 g/dL (NR: 12 - 15 g/dL), erythrocyte sedimentation rate (ESR) 6 mm/h (NR: 0 - 19 mm/h), platelets 413 × 103/µL (NR: 150 - 400 × 103/µL), creatinine 0.47 mg/dL (NR: 0.5 - 0.9 mg/dL), urea 12 mg/dL (NR: 10 - 50 mg/dL), total bilirubin 0.36 mg/dL (NR: 0.2 - 1.0 mg/dL), aspartate transaminase 33 U/L (NR: 10 - 30 U/L), alanine transaminase 33 U/L (NR: 10 - 36 U/L), alkaline phosphatase 34 U/L (NR: 32 - 104 U/L), gamma-glutamyltransferase 168 U/L (NR: 6 - 39 U/L), albumin 4.06 mg/dL (NR: 3.5 - 5.0 g/dL) and C-reactive protein (CRP) 11 mg/L (NR: 0.0 - 5.0 mg/L).
Diagnostic paracentesis was performed (50 mL of ascitic fluid), whose cytochemical analysis showed proteins 4.40 g/dL (NR: 0.30 - 4.10 g/dL), albumin 3.00 g/dL, serum ascites albumin gradient < 1.1, lactate dehydrogenase (LDH) 153 U/L, red blood cells 5,120/µL (NR: 0 - 100,000/µL), WBC 3,680/µL (NR: 0 - 500/µL), eosinophils 2,400/µL, normal adenosine deaminase (13 U/L, NR: 0 - 20 U/L). No microbiological growth in the peritoneal fluid was detected, including mycobacteria.
The patient suffered worsening of complaints in the first 48 h, with profuse diarrhea, severe abdominal pain, and AEC rising (5.23 × 103/µL). She was put on systemic corticosteroid therapy with a single dose of 125 mg intravenous (IV) methylprednisolone and then 40 mg prednisolone with immediate AEC reduction (Fig. 1), progressive improvement of abdominal complaints, reduction in the number of bowel discharges, and regression of ascites without the need to drain.
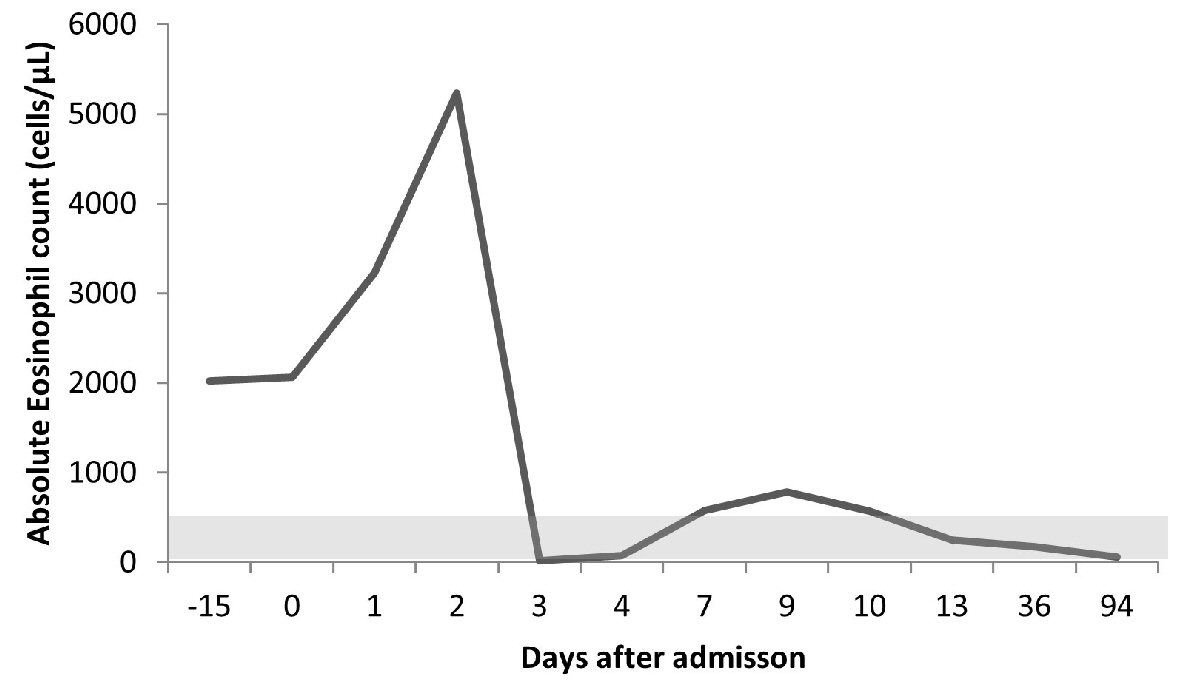 Click for large image | Figure 1. Evolution of the peripheral blood absolute eosinophil count (normal range (light-gray): 40 - 400 cells/µL) during the disease course. Day 0 refers to admission day. The patient started corticosteroids on day 2 and was discharged on day 10. Thereafter, the patient was observed as an outpatient on days 13, 36, and 94, remaining stable despite progressive corticosteroid tapering. |
The etiologic study of the systemic eosinophilia showed (Table 1): normal IgG (1,061 mg/dL, NR: 793 - 1,590 mg/dL) and an elevated IgG4 (183.0 mg/dL, NR: 7 - 89 mg/dL); without hypocomplementemia; negative anti-nuclear antibodies (ANAs), anti-double stranded DNA (anti-dsDNA), anti-Sm, anti-SSa, anti-SSb, antineutrophil cytoplasmic antibodies (ANCAs), and anti-Saccharomyces cerevisiae antibody (ASCA); normal angiotensin-converting enzyme (22 U/L, NR: 8 - 76 U/L) and serum protein electrophoresis without monoclonal peaks. Normal values of β2-microglobulin (1.61 mg/L, NR: < 2.53 mg/L), tryptase (2.41 µg/L, NR: < 11.4 µg/L), vitamin-B12 (276.9 pg/mL, NR: 191 - 663 pg/mL)) were also observed. Serological studies were negative for syphilis, human immunodeficiency virus (HIV), human T-cell lymphotropic virus (HTLV) I and II, hepatitis A and C virus. The patient was vaccinated for hepatitis B.
 Click to view | Table 1. Immunology and Serology Laboratory Results of the Young Female Patient |
Stool analysis revealed fat droplets and some digested muscle fibers, chymotrypsin 48.0 U/g stool (NR > 13.2 U/g), fecal calprotectin 384 µg/g stool (NR: < 50 µg/g; value > 200 µg/g indicates severe gastrointestinal inflammation), and positive occult blood test. Parasitological examination and search for Salmonella, Campylobacter, Escherichia coli (E. coli) 0157, Yersinia, and Giardia lamblia in feces were negative. Serological studies for Fasciola hepatica, Toxocara canis, Schistosoma, Echinococcus, Ascaris, and Strongyloides stercoralis were also negative. Upper digestive endoscopy and ileocolonoscopy were performed 2 days after starting corticosteroids, without evidence of macroscopic lesions. The histological analysis of the random biopsies performed (Figs. 2, 3) revealed the presence of light inflammatory infiltrate in the esophagus composed mainly by eosinophils (Fig. 2); stomach with signs of superficial gastritis; duodenum with a polymorphic inflammatory infiltrate in the chorion with frequent eosinophils; ileus with discrete to moderate polymorphic inflammatory infiltrate in the chorion containing frequent eosinophils expanding the lamina propria, signs of activity and images of transepithelial migration of lymphocytes (Fig. 3); and colon with a slight distortion of the architecture and decreased mucosecretory activity, moderate to intense polymorphic inflammatory infiltrate, containing eosinophils, expanding the lamina propria, subepithelial lymphoid aggregates, and absence of granulomas.
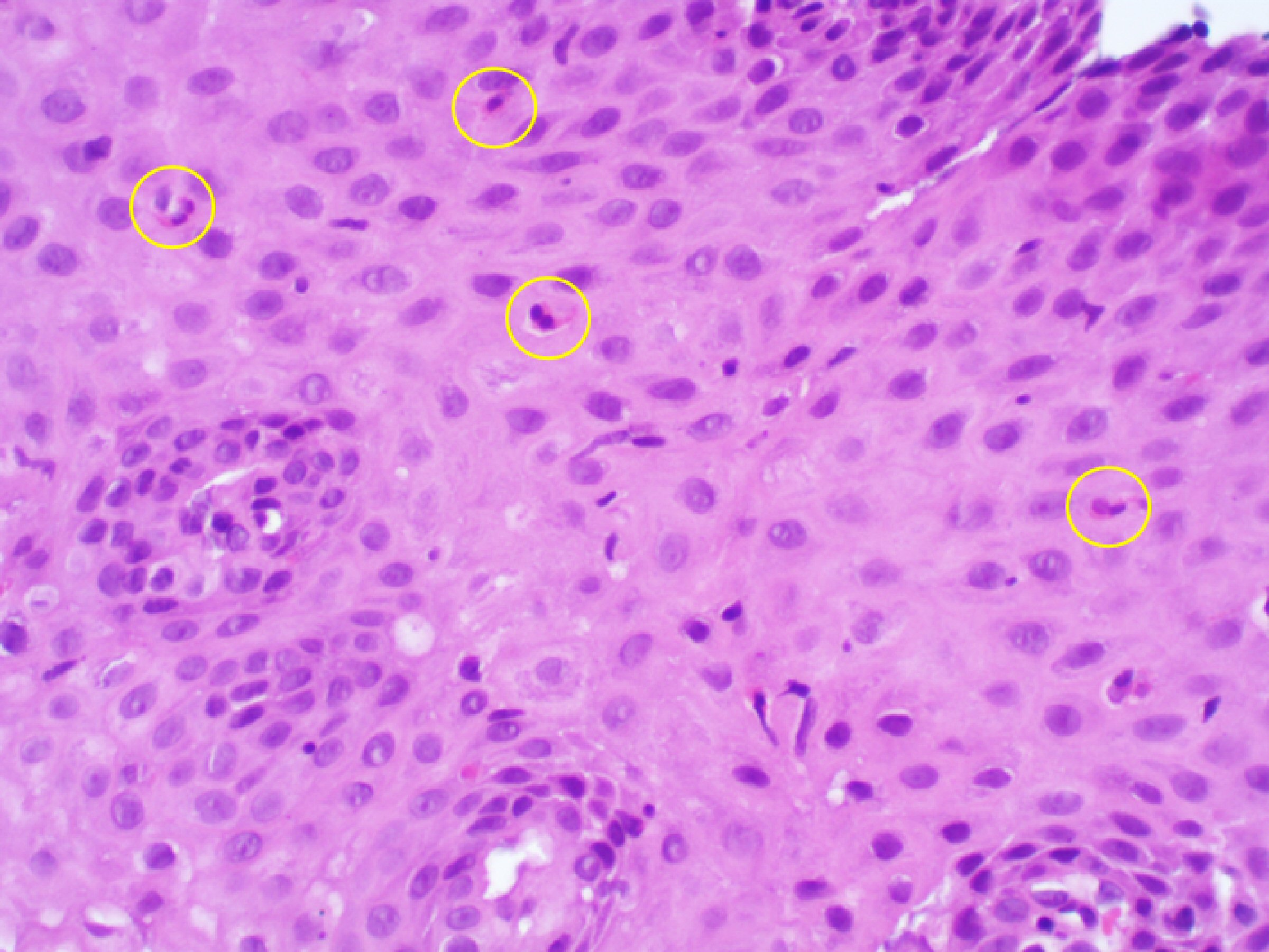 Click for large image | Figure 2. Upper digestive tract of the young female patient 2 days after starting the steroid therapy. In the superficial esophagus biopsy fragments obtained in the upper digestive tract endoscopy there was a light inflammatory infiltrate composed mainly by eosinophils, recognizable by their cytoplasmic granules that appear brick-red after eosin staining (yellow circles) with a maximum of five eosinophils per high-power field. (hematoxylin and eosin stain (H&E), × 200 magnification). |
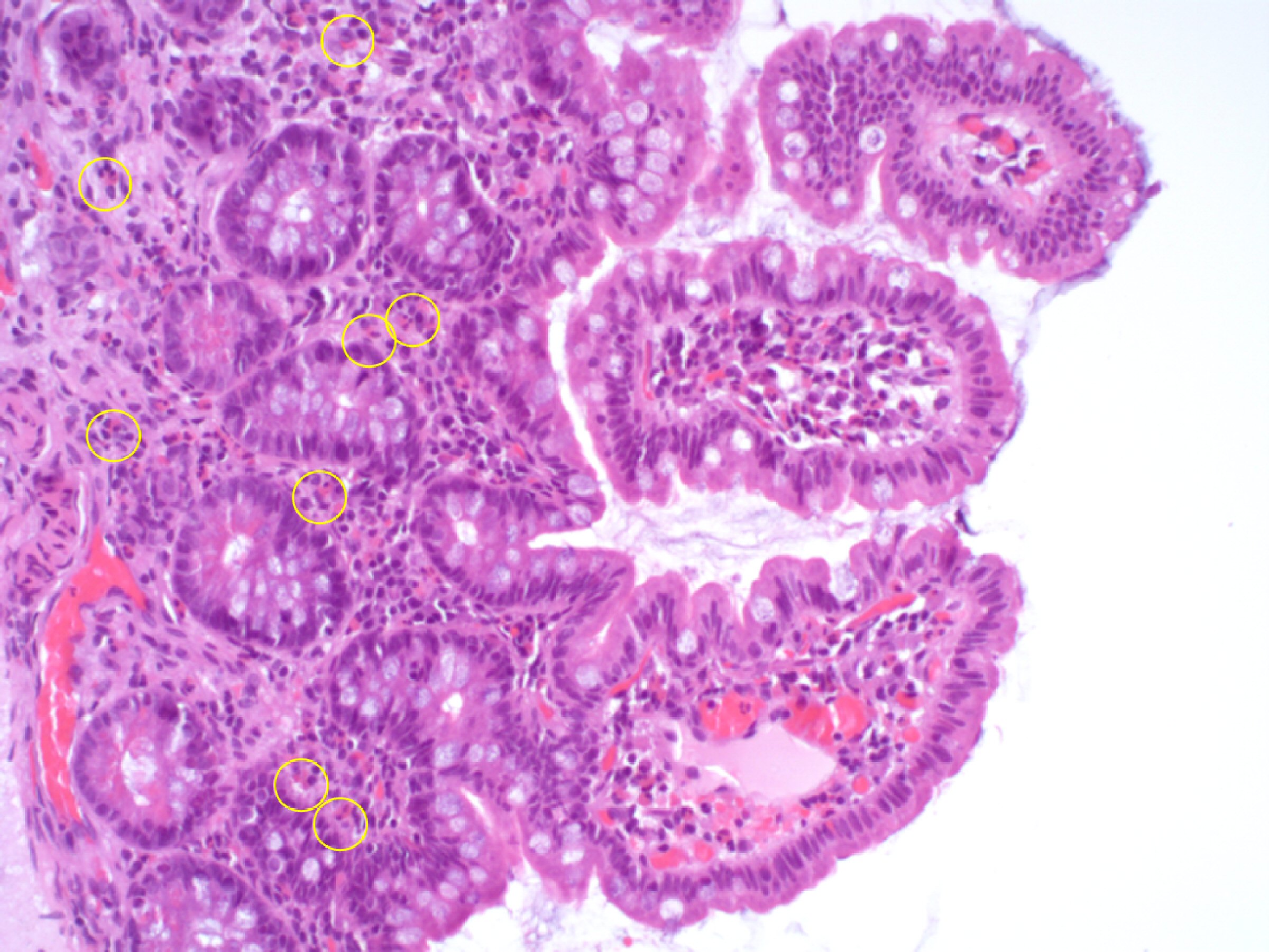 Click for large image | Figure 3. Lower digestive tract of the young female patient 2 days after starting methylprednisolone. In the ileal biopsy fragments obtained in the lower digestive tract endoscopy the global architecture and mucosecretory activity were globally preserved, with the chorion containing a mild polymorphic inflammatory infiltrate rich in eosinophils, recognizable by their cytoplasmic granules that appear brick-red after eosin staining (yellow circles) (hematoxylin and eosin stain (H&E), × 200 magnification). |
Bone marrow (BM) aspirate was performed 7 days after starting corticosteroids, showing normocellular BM, myeloid series without morphological changes, eosinophils, and their precursors with normal morphology, small lymphocytes and condensed chromatin, no suggestive signs of involvement by neoplastic cells. Search for FIP1L1/PDGFRa transcripts using nested RT-PCR was negative. Peripheral blood cytometry showed no changes pointing to lymphoproliferative disease.
On the eighth day of corticosteroid therapy, the abdominal CT scan showed slightly increased homogeneous liver with regular contours and without focal nodular changes; pancreas with globose morphology, diffuse swelling in the body and tail suggestive of autoimmune pancreatitis; multiple mesenteric infracentimetric adenopathies; and small volume ascites. No other relevant findings were observed. A pancreatic biopsy was not performed.
Autoimmune pancreatitis in the context of IgG4-RD was assumed with secondary HES with enterocolitis and eosinophilic ascites. The patient was discharged after 10 days with tapering corticosteroids. Six months later, the patient is being followed as an outpatient. Her condition is stable, without significant symptoms, and she is taking prednisolone 2.5 mg once daily (OD).
| Discussion | ▴Top |
IgG4-RD is a multisystemic immune-mediated fibroinflammatory disease [1] that can involve the pancreas as an AIP [3]. Regarding AIP criteria (HISORt) [4], our patient fulfills three diagnostic criteria: pancreatic imaging suggestive of autoimmune pancreatitis, serology criteria with IgG4 ≥ 2 times the normal upper limit, and response to steroid treatment with marked improvement pancreatic and extrapancreatic manifestations. The histological criterion was not accessed since pancreatic biopsy was not performed, as it is an invasive procedure; it was not necessary to assume the diagnosis and the patient had a favorable response to treatment. The involvement of the gastrointestinal tract in IgG4-RD may occur, but it is uncommon [3].
HES is a rare syndrome with hypereosinophilia (> 1.5 × 103/µL) and organ damage [5] that can be primary (myeloproliferative), secondary or unknown, being IgG4-RD a secondary cause of HES [8]. In contrast to primary HES, the eosinophilia secondary to IgG4-RD is usually mild to moderate, typically quite evanescent, ablated by steroids or rituximab therapy [6]. In our patient’s case, who presented HES, primary HES was the main differential diagnosis; however, the absence of alterations of the myeloid lineage in BM, negative FIP1L1/PDGFRa gene fusion, and a positive response after starting corticosteroid therapy does not corroborate this diagnosis.
Another diagnosis that was considered was ANCA-associated vasculitis, however, it was discarded due to the absence of findings in the immunological study, absence of granulomas, and the absence of elevation of CRP/ESR that usually accompany vasculitis.
Thus, the diagnosis of IgG4-RD was assumed with secondary HES with organ damage at the gastrointestinal tract and ascites.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Both written and verbal informed consent were obtained from the patient for publication of this case report.
Author Contributions
LSP collected the case data, wrote the manuscript, and prepared the table and the pictures; NJL collected the case data, prepared the pictures (pathology), and wrote the manuscript; AC, AF and ARC collected the case data and revised the manuscript. All authors approved the final version to be published.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Wallace ZS, Naden RP, Chari S, Choi HK, Della-Torre E, Dicaire JF, Hart PA, et al. The 2019 American College of Rheumatology/European League Against Rheumatism classification criteria for IgG4-related disease. Ann Rheum Dis. 2020;79(1):77-87.
doi pubmed - Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, Kloppel G, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25(9):1181-1192.
doi pubmed - Vashi B, Khosroshahi A. IgG4-related disease with emphasis on its gastrointestinal manifestation. Gastroenterol Clin North Am. 2019;48(2):291-305.
doi pubmed - Chari ST, Takahashi N, Levy MJ, Smyrk TC, Clain JE, Pearson RK, Petersen BT, et al. A diagnostic strategy to distinguish autoimmune pancreatitis from pancreatic cancer. Clin Gastroenterol Hepatol. 2009;7(10):1097-1103.
doi pubmed - Carruthers MN, Park S, Slack GW, Dalal BI, Skinnider BF, Schaeffer DF, Dutz JP, et al. IgG4-related disease and lymphocyte-variant hypereosinophilic syndrome: A comparative case series. Eur J Haematol. 2017;98(4):378-387.
doi pubmed - Chen LYC, Mattman A, Seidman MA, Carruthers MN. IgG4-related disease: what a hematologist needs to know. Haematologica. 2019;104(3):444-455.
doi pubmed - Chen C, Chen K, Huang X, Wang K, Qian S. Concurrent eosinophilia and IgG4-related disease in a child: A case report and review of the literature. Exp Ther Med. 2018;15(3):2739-2748.
doi pubmed - Klion AD. How I treat hypereosinophilic syndromes. Blood. 2015;126(9):1069-1077.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.