

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 12, Number 9, September 2021, pages 339-342

Infection as Trigger for Congenital Thrombotic Thrombocytopenic Purpura in an Adult Patient
Diego Alberto Lozano Jaramilloa, b, Marco Alejandro Jimenez Ochoaa
aCentro Medico Nacional Siglo XXI Instituto Mexicano del Seguro Social, Mexico City, Mexico
bCorresponding Author: Diego Alberto Lozano Jaramillo, Centro Medico Nacional Siglo XXI Instituto Mexicano del Seguro Social, Avenida Cuauhtemoc 300, Colonia Doctores, Postal Code 06720, Mexico City, Mexico
Manuscript submitted June 18, 2021, accepted July 3, 2021, published online August 25, 2021
Short title: Infection as Trigger in an Adult cTTP Case
doi: https://doi.org/10.14740/jmc3737
| Abstract | ▴Top |
Congenital thrombotic thrombocytopenic purpura (cTTP) is an inherited disease that is sometimes fatal in early childhood. cTTP is similar to idiopathic thrombotic thrombocytopenic purpura (iTTP); both are characterized by varying levels of thrombocytopenia, microangiopathic hemolytic anemia (MAHA), and end-organ damage secondary to occlusion of the microvasculature. cTTP is caused by a partial or total deficiency or loss of function of ADAMTS-13 (a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13). We report the case of a 33-year-old woman who was mistakenly diagnosed with primary immune thrombocytopenia (ITP) during childhood. The patient was referred to our center with dyspnea, fatigue, fever, and jaundice with no clinical bleeding. Laboratory features were compatible with MAHA; ADAMTS-13 activity was at 0%, with negativity for ADAMTS-13 antibodies. We concluded the final diagnosis was cTTP. The triggering factor identified for MAHA was a double infection: central venous catheter bacterial infection and atypical pneumonia. After 7 days of treatment with antibiotics and ongoing total plasma exchange (TPE), the patient responded favorably. Our patient received fresh frozen plasma (FFP) infusion once every 2 weeks, and prophylactic voriconazole remained under control at the time of writing. As demonstrated in this case, effective treatment of the trigger cause helps reduce the need for continuous FFP exposure and controls the MAHA.
Keywords: Congenital thrombotic thrombocytopenic purpura; Fresh frozen plasma; Microangiopathic hemolytic anemia; ADAMTS-13; Total plasma exchange
| Introduction | ▴Top |
Congenital thrombotic thrombocytopenic purpura (cTTP), also known as Upshaw-Schulman syndrome, is a rare disease usually diagnosed during childhood [1]. cTTP features include thrombocytopenia, microangiopathic hemolytic anemia (MAHA), and end-organ damage [2]. cTTP is caused by a wide variety of mutations that alters the function or synthesis of ADAMTS-13 (a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13). These mutations can range from compound heterozygous or homozygous, with various clinical presentations related to the severity of ADAMTS-13 deficiency or levels of functional enzyme [3]. In Mexico and Latin America, there is no reported incidence or prevalence of this disease.
| Case Report | ▴Top |
Investigations
A 33-year-old woman was referred to our hospital with a history of neonatal jaundice and was treated with phototherapy; and no specific diagnosis at that time was made. During early childhood (2 years old), primary immune thrombocytopenia (ITP) was diagnosed based on low platelet counts and occasional epistaxis. Treatment with corticosteroids was administered, but the response was not adequately noted. Splenectomy was performed at 9 years of age with no apparent complications. She never achieved complete remission after surgery and was corticosteroid dependent. She also had a history of mild renal disease without the need for renal replacement therapy (RTT), primary hypothyroidism controlled with levothyroxine, hepatitis C virus (HCV) infection in response to second-line therapy with a sustained virologic response at the time of referral. On admission to our hospital, she presented anemic syndrome characterized by dyspnea, fatigue, and tachycardia, accompanied by jaundice, with no clinical signs of ongoing bleeding or purpura.
Diagnosis
Laboratory features included a platelet count of 19.0 × 103/µL, hemoglobin 6.0 g/dL, mean corpuscular volume of 97.9 fL, lactate dehydrogenase (LDH) level of 2,950 U/L, creatinine 3.51 mg/dL, total bilirubin 1.54 mg/dL and indirect bilirubin 0.93 mg/dL. The blood film revealed abundant schistocytes and microspherocytes. ADAMTS-13 activity levels were 0%, and ADAMTS-13 antibodies were negative, supporting the diagnosis of cTTP. Activity levels were measured again 3 months later and were still at 0%.
Treatment
Total plasma exchange (TPE) was performed. Response was achieved after the ninth procedure. The patient was discharged from the hospital with no significant complications, and central venous catheter was removed.
A few weeks later, she began with epistaxis, fever, and laboratory features compatible with MAHA despite adequate prophylaxis with fresh frozen plasma (FFP) infusion. A new central venous catheter was placed and TPE was ordered daily, but no response was observed after the fifth procedure. A central venous catheter blood culture was performed, and Acinetobacter baumannii was identified. Treatment began with cefepime for 14 days and removal of the catheter. Despite these actions still, no response was noted. TPE was continuously being performed. Days later, she began with sudden dyspnea and a persistent fever up to 38.8 °C. A pulmonary computed tomography (CT) scan was performed (Fig. 1). The image suggested atypical pneumonia, so voriconazole, linezolid, and imipenem were initiated empirically, and a polymerase chain reaction (PCR) for coronavirus disease 2019 (COVID-19) was issued. PCR test was negative. Bronchoscopy was not possible to perform due to initial suspicion of COVID-19 infection. Subsequent blood cultures Klebsiella pneumoniae was identified. Sputum cultures had no growth in 72 h. After 7 days of treatment and continuous TPE improvement was noted. The hemoglobin raised to 8.4 g/dL, platelet to 418 × 103/µL, and LDH levels lowered to 331 U/L (Table 1). The patient was discharged with prophylactic voriconazole after achieving a complete response.
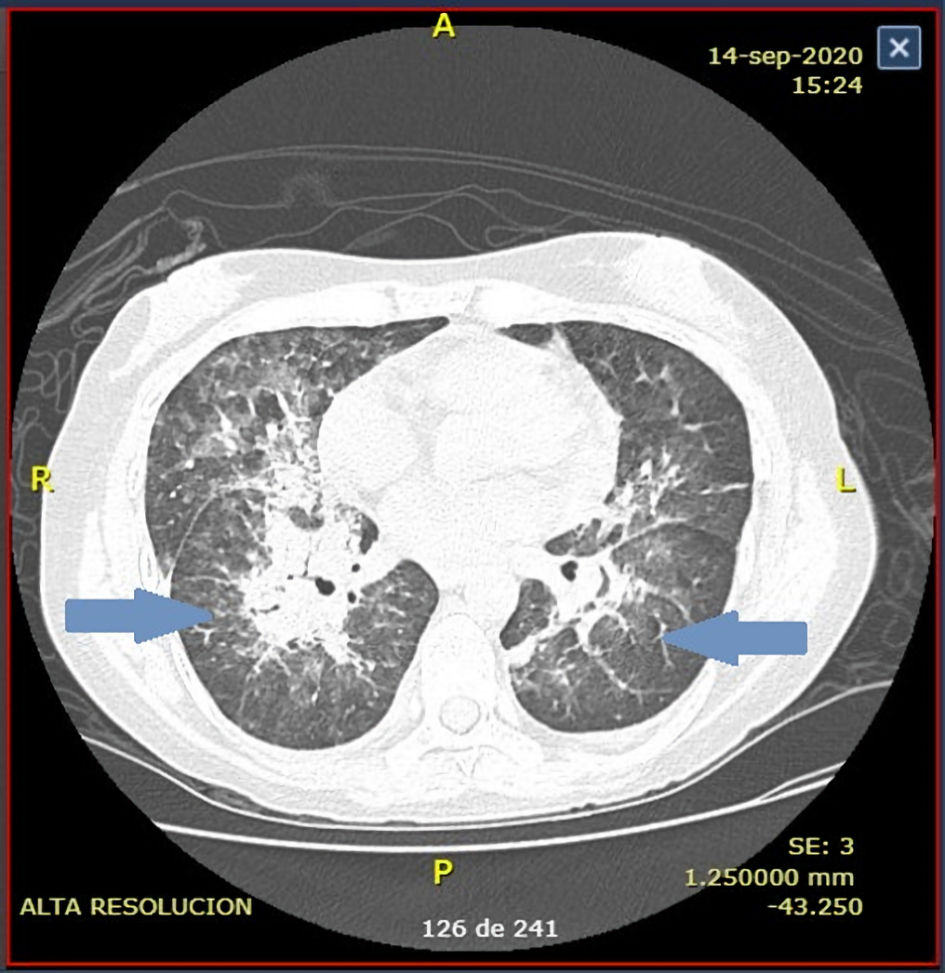 Click for large image | Figure 1. Chest computed tomography (CT) scan at presentation reveals diffuse nodular interstitial infiltrates in the upper lobules. |
 Click to view | Table 1. Laboratory Features |
Follow-up
A new CT scan was performed a month later with clear radiological and clinical improvement (Fig. 2). Today, she receives FFP infusion every 2 weeks, calculated at 15 mL/kg with stable clinical response and no TPE need.
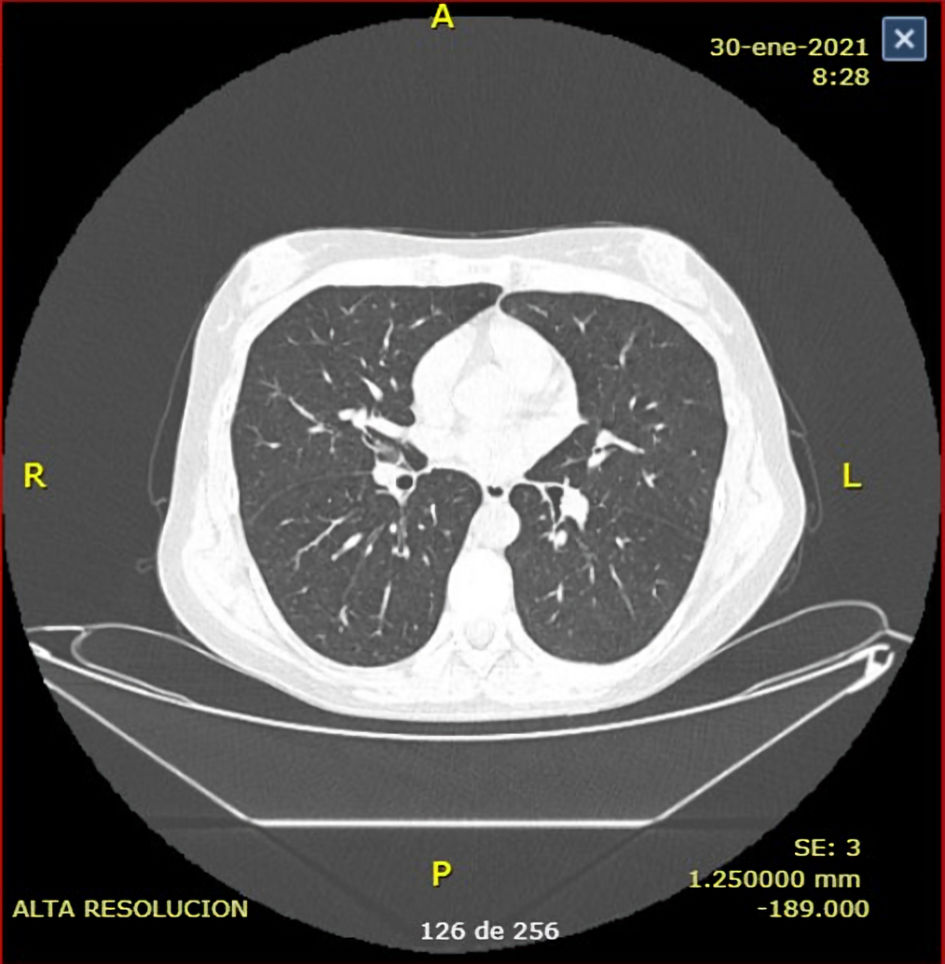 Click for large image | Figure 2. Chest computed tomography (CT) scan after treatment reveals normal parenchyma without signs of significant fibrosis. |
| Discussion | ▴Top |
cTTP is an exceedingly rare disease, even more than idiopathic thrombotic thrombocytopenic purpura (iTTP). The estimated prevalence worldwide is unknown but is estimated to be around 16.7 cases per million [4]. Historically iTTP was described as a pentad, compromising microangiopathic hemolytic anemia, fever, neurological symptoms, renal impairment, and thrombocytopenia; but cTTP varies widely in its forms of presentation [1]. It can range from a severe disease diagnosed in early childhood to a mild disease diagnosed in adults that is classically mistaken for another cause of thrombocytopenia, most commonly ITP like in our case we reported. The approach we took considered a triggering factor as the MAHA cause and ultimately led to the correct diagnosis of cTTP in our patient. We currently do not have the means to perform genetic studies to determine the responsible mutation, which is our approach’s main limitation.
Some clinical manifestations are more prominent in cTTP than in iTTP, and may help us differentiate between both. For example, it is described that patients with cTTP have a higher risk of transient ischemic attack (TIA), occurring mainly in young adults, not noted in our patient. In the International Hereditary Thrombotic Thrombocytopenia Purpura Registry, up to 4% had an ITA [5]. Also, chronic kidney injury is reported more frequently in cTTP. It was reported as high as 9% in patients with acute kidney injury at 2 years of age [5]. It is estimated that only 10% who developed chronic kidney injury will need dialysis [5]. Our reported case did not need dialysis at the moment of admission or after controlling the MAHA.
The severity of the disease correlates with the underlying mutation. More than 200 ADMATS-13 mutations have been implicated in these patients [3, 7]. Some of them have increased frequency and vary extensively with each geographic region. The two most common mutations in European ancestry are the p.R1060W and the insertion c.4143_4144dupA [6]. There are no studies in Latin American that determine the most common mutations affecting the ADAMTS-13 gene.
The usual time of presentations varies from patient to patient. Typically, if presenting in young adult women, the need for pregnancy, alcohol, or drug use needs to be identified as triggering factors [6]. In many patients, there is no identifiably trigger [7]. Infections are known as common trigger causes. There are no studies that describe the most common microorganisms that promote this. In our case, a bacterial infection of the central venous catheter where TPE was performed and atypical pneumonia was the identified causes. There are no case reports of atypical pneumonia as a trigger for the disease, but it is known that potentially any infection can lead to it [8].
Atypical pneumonia was diagnosed and treated empirically, utilizing the current guidelines as in non-immunocompromised patients [9]. Our patient may be immunocompromised as she was status post splenectomy, and she was not using steroid treatment at the time of admission. It is not known if patients with cTTP carry a predisposition to any infection.
The optimal treatment for the MAHA is not well defined. FFP infusions are typically reserved for patients who are in complete remission as prophylactic treatment. Current International Society of Thrombosis and Hemostasis (ISTH) guidelines recommend infusion every 1 - 3 weeks at a dose of 10 - 15mL/kg body weight as prophylaxis [10]. TPE has been reported in the literature as an effective emergency treatment, but it is not a viable option in the prophylactic setting because of logistic reasons and the increased exposure to FFP donors. One retrospective study by Kovarova et al concluded that TPE is an option for prophylaxis that can prolong the need for FFP infusion (21 vs. 14 days of the need of new TPE or FFP infusion, respectively), although there were no differences in ADAMTS-13 activity immediately after these two procedures (P > 0.5) [11]. There are currently no clinical trials comparing both treatments.
A promising new treatment is recombinant ADAMTS-13. A phase 1 study was performed on 15 patients with severe ADAMTS-13 (defined as an activity less than 6%). After a single dose of 5, 20, or 40 U/kg body weight of recombinant ADAMTS-13, the study demonstrated pharmacokinetic properties comparable to plasma infusion with a superior safety profile because no patients exhibited anaphylaxis or allergic reactions [12].
cTTP continues to be a diagnostic challenge in the world, especially in Latin America. Not all centers have the means necessary to identify ADAMTS-13 activity, ADAMTS-13 antibodies, and the mutation affecting the ADAMTS-13 gene. These limitations explain the lack of epidemiological data. There is no doubt that more studies are needed to determine the most optimal treatment for the MAHA in patients with cTTP. It seems that prophylactic plasma infusion is a good and inexpensive option.
Learning points
Attention needs to be put to the triggering factors because if not treated, the MAHA will persist even FFP infusion or TPE is performed. Additionally, it seems that recombinant ADAMTS-13 is the future therapy for treating and preventing MAHA in cTTP patients without the hazard of exposing patients to FFPs. It remains to be seen if it is a safe treatment with a concomitant infection.
Acknowledgments
We would like to acknowledge all the residents of hematology of our center who were involved in the well-being of this patient.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Informed consent was waved because the manuscript was made according to the current standard, and the patient identification was kept secret for protection purposes.
Author Contributions
MAJO and DALJ both wrote and edited the manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Kremer Hovinga JA, Braschler TR, Buchkremer F, Farese S, Hengartner H, Lovey PY, Largiader CR, et al. Insights from the hereditary thrombotic thrombocytopenic purpura registry: discussion of key findings based on individual cases from Switzerland. Hamostaseologie. 2020;40(S 01):S5-S14.
doi pubmed - Kremer Hovinga JA, Heeb SR, Skowronska M, Schaller M. Pathophysiology of thrombotic thrombocytopenic purpura and hemolytic uremic syndrome. J Thromb Haemost. 2018;16(4):618-629.
doi pubmed - Fujimura Y, Matsumoto M, Isonishi A, Yagi H, Kokame K, Soejima K, Murata M, et al. Natural history of Upshaw-Schulman syndrome based on ADAMTS13 gene analysis in Japan. J Thromb Haemost. 2011;9(Suppl 1):283-301.
doi pubmed - von Krogh AS, Quist-Paulsen P, Waage A, Langseth OO, Thorstensen K, Brudevold R, Tjonnfjord GE, et al. High prevalence of hereditary thrombotic thrombocytopenic purpura in central Norway: from clinical observation to evidence. J Thromb Haemost. 2016;14(1):73-82.
doi pubmed - van Dorland HA, Taleghani MM, Sakai K, Friedman KD, George JN, Hrachovinova I, Knobl PN, et al. The international hereditary thrombotic thrombocytopenic purpura registry: key findings at enrollment until 2017. Haematologica. 2019;104(10):2107-2115.
doi pubmed - Lotta LA, Garagiola I, Palla R, Cairo A, Peyvandi F. ADAMTS13 mutations and polymorphisms in congenital thrombotic thrombocytopenic purpura. Hum Mutat. 2010;31(1):11-19.
doi pubmed - Veyradier A, Meyer D, Loirat C. Desmopressin, an unexpected link between nocturnal enuresis and inherited thrombotic thrombocytopenic purpura (Upshaw-Schulman syndrome). J Thromb Haemost. 2006;4(3):700-701.
doi pubmed - Kremer Hovinga JA, George JN. Hereditary thrombotic thrombocytopenic purpura. N Engl J Med. 2019;381(17):1653-1662.
doi pubmed - Limper AH, Knox KS, Sarosi GA, Ampel NM, Bennett JE, Catanzaro A, Davies SF, et al. An official American Thoracic Society statement: Treatment of fungal infections in adult pulmonary and critical care patients. Am J Respir Crit Care Med. 2011;183(1):96-128.
doi pubmed - Zheng XL, Vesely SK, Cataland SR, Coppo P, Geldziler B, Iorio A, Matsumoto M, et al. ISTH guidelines for treatment of thrombotic thrombocytopenic purpura. J Thromb Haemost. 2020;18(10):2496-2502.
doi pubmed - Kovarova P, Hrdlickova R, Blahutova S, Cermakova Z. ADAMTS13 kinetics after therapeutic plasma exchange and plasma infusion in patients with Upshaw-Schulman syndrome. J Clin Apher. 2019;34(1):13-20.
doi pubmed - Scully M, Knobl P, Kentouche K, Rice L, Windyga J, Schneppenheim R, Kremer Hovinga JA, et al. Recombinant ADAMTS-13: first-in-human pharmacokinetics and safety in congenital thrombotic thrombocytopenic purpura. Blood. 2017;130(19):2055-2063.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.