

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 12, Number 11, November 2021, pages 455-459

Dilated Cardiomyopathy due to the Novel MT-CYB Missense Mutation m.14757T>C
Sinda Zarrouka, Josef Finstererb, d, Sounira Mehria, Fatma Ourdac, Saida Ben Araba, Raafik Boussadac
aDepartment of Genetic and Molecular Epidemiology, Medical University of Tunis, Tunisia
bCity Hospital Landstrasse, Messerli Institute, Vienna, Austria
cDepartment of Functional Cardiology, La Rabta Hospital of Tunis, Tunisia
dCorresponding Author: Josef Finsterer, City Hospital Landstrasse, Vienna, Austria
Manuscript submitted September 10, 2021, accepted October 13, 2021, published online November 5, 2021
Short title: Novel Missense Mutation in DCM
doi: https://doi.org/10.14740/jmc3787
| Abstract | ▴Top |
Mitochondrial DNA (mtDNA) mutations frequently manifest with multisystem disease, including cardiomyopathy (CM). Various studies described mutations in protein-encoding mtDNA genes, such as cytochrome-b, manifesting with CM. A detailed clinical, biochemical, and molecular genetic analysis was performed in a 40-year-old male with dilated CM (DCM) to detect the underlying mtDNA defect. Muscle biopsy showed complex-III deficiency, and sequencing of the cytochrome-b gene revealed the pathogenic variant m.14757T>C in MT-CYB, resulting in the replacement of the hydrophobic methionine by the polar threonine (M4T). By application of the PolyPhen algorithm the variant was predicted as pathogenic. The mutation was not found in 100 healthy controls and never reported as a neutral polymorphism despite extensive sequencing of the cytochrome-b gene in 2,704 normal healthy controls from different ethnic backgrounds. In conclusion, the novel variant m.14757T>C in MT-CYB is associated with DCM suggesting a pathophysiologic role of the variant in the development of DCM.
Keywords: Cytochrome-b gene; Dilated cardiomyopathy; Systolic dysfunction; Mitochondria; Oxidative phosphorylation; mtDNA; Mitochondrial
| Introduction | ▴Top |
Dilated cardiomyopathy (DCM) is characterized by widening of the cardiac cavities, reduced systolic function, and normal coronary angiography [1]. DCM is frequently associated with heart failure, cardiac rhythm abnormalities and intra-ventricular thrombus formation. If heart failure is intractable to drug therapy, heart transplantation is indicated. The particular pathophysiological mechanism leading to ventricular dilatation and systolic dysfunction is poorly comprehended. During the last few years, however, attention has been focused on abnormalities of contractile and structural myocardial proteins and on disorders of the mitochondrial energy metabolism [1]. A number of CMs were attributed to abnormal respiratory chain functions, particularly those caused by mitochondrial DNA (mtDNA) mutations [2-4].
The heart contains abundant mitochondria, which constitute approximately 40% of total cardiomyocyte volume [5]. Their major function is the provision of energy in terms of adenosine triphosphate (ATP) by the oxidative phosphorylation system (OXPHOS). ATP is required by the actomyosin adenosine triphosphatase (ATPase) for contraction and relaxation of cardiomyocytes. Additionally, membrane-bound ATPase utilizes ATP for depolarization and repolarization of cell membranes. Decreased ATP levels may thus not only impair contractility but may also lower the mitochondrial membrane potential and thus impair impulse generation and conduction along the cardiac conduction system, manifesting as arrhythmias [6]. Energy production in mitochondria involves five oligomeric complexes which are embedded within the inner mitochondrial membrane. Bc1 is one of these complexes and composed of three catalytic subunits (cytochrome-b, cytochrome-c1, and the Rieske iron-sulfur protein) and seven or eight non-catalytic subunits [6]. Cytochrome-b plays a crucial role for the activity of the bc1 complex since it harbors two b-hemes and determines the shape of the two ubiquinone fixation sites. It is the only subunit of bc1 encoded by an mtDNA located gene. The cytochrome-b gene (MT-CYB) is located at nucleotide position 14747-15887, and is flanked by the tRNA genes for glutamic acid and threonine. It encodes a protein of 380 amino acids (AAs) [7]. Mutations in MT-CYB resulting in decreased complex-III-activity have been frequently described in humans [8] and experimental models [9-13] of CM or heart failure. In order to elucidate the cause of DCM in a patient with a complex-III defect, MT-CYB was screened for pathogenic mutations in the proband and 100 healthy controls.
| Case Report | ▴Top |
Investigations
The patient is a 40-year-old male in whom DCM was diagnosed upon clinical, echocardiographic, and angiographic investigations, carried out in the Department of Cardiology, Rabta University Hospital, Tunis, Tunisia. Transthoracic echocardiography revealed a left ventricular end-diastolic diameter (LVEDD) of 65 mm; a fractional shortening (FS) of 23%; and an ejection fraction (EF) of 48%. Coronary angiography was normal. The control group comprised 100 healthy adult subjects. The study was approved by the Ethical Committee of the University of Tunis (SN745556).
Diagnosis
Muscle histopathological and biochemical studies
Muscle specimens were obtained by open biopsy, frozen, and stored in liquid nitrogen. Biochemical measurement of the ubiquinone oxidoreductase (NADH) dehydrogenase (complex-I), succinate-cytochrome-c-reductase (complex-II and complex-III), cytochrome-c-oxidase (complex-IV) and citrate synthase activities were done as described previously [14].
Cytochrome-b gene analysis
Total DNA was extracted from lymphocytes or from muscle tissue according to established methods. The fragment that overlapped the entire sequence of MT-CYB (14747-15887) was attained by using a polymerase chain reaction (PCR) assay with cytb-1F as the first forward primer (nucleotides 14667-14687), cytbTB-1R as the first reverse primer (nucleotides 15289-15269), cytb-2F as the second forward primer (nucleotides 15189-15209) and cytb-2R as the second reverse primer (nucleotides 15941-15921). PCR amplification was performed in a 50 volume containing 100 ng DNA, 10 pmol of each primer, 4 µL MgCl2, 5 µL dNTP, and 0.2 µL Taq polymerase. Conditions for DNA fragment amplification included 3 min at 94 °C, 30 cycles of 30 s at 94 °C, 60 s at 52 °C, 1 min 30 s at 72 °C, and 10 min at 72 °C.
Direct sequencing of PCR-amplified fragments
Direct sequencing of PCR fragments was conducted using an ABI 3730 automated DNA sequencer (Applied Biosystems) by using a Big Dye Terminator V.3.1 cycle sequencing kit (Applied 4337455 (100 reactions)) according to the manufacturer’s recommendations. All sequences were numbered according to the L-strand (the coding strand) of the Cambridge reference sequence [7].
Prediction data
Mutated protein structures change due to biochemical differences of the AA variants (acidic, basic, hydrophobic) or due to the variant changing the protein sequence and affecting the tertiary or quaternary structure or the active site. To distinguish deleterious from functionally neutral mutations we used the PolyPhen algorithm, which predicts the pathogenicity of a specific variant by using a set of empirical rules, based on sequence, phylogenetic and structural information characterizing the variant. In addition to sequence alignments, PolyPhen utilises protein structure databases, such as the Protein Data Bank (PDB), Protein Quarternary Structure (PQS), Dictionary of Secondary Structure in Proteins (DSSP), or three-dimensional structure databases to assess if a variant affects the protein’s secondary structure, the interchain contacts, functional sites, or the binding sites.
Biochemical studies of the muscle homogenate revealed low activities of complex-II and -III but virtually normal complex-I and complex-IV activities (Table 1) indicating a complex-III defect. Molecular analyses of muscle and lymphocyte mtDNA revealed the transition thymine for cytosine at np14757 (Fig. 1), changing medium size and hydrophobic methionine (M) to medium size and polar (hydrophilic) threonine (T) (M4T). This mutation was not found in lymphocyte mtDNA of 100 healthy subjects without comorbidities or known genetic defect. A substitution of adenine for guanine at np15326, changing medium size and hydrophilic threonine (T) to small size and hydrophobic alanine (A) (T194A), was considered a neutral polymorphism based on previous reports [15].
 Click to view | Table 1. Activities of Respiratory Chain Enzymes in the Muscle Extract of the Described Patient |
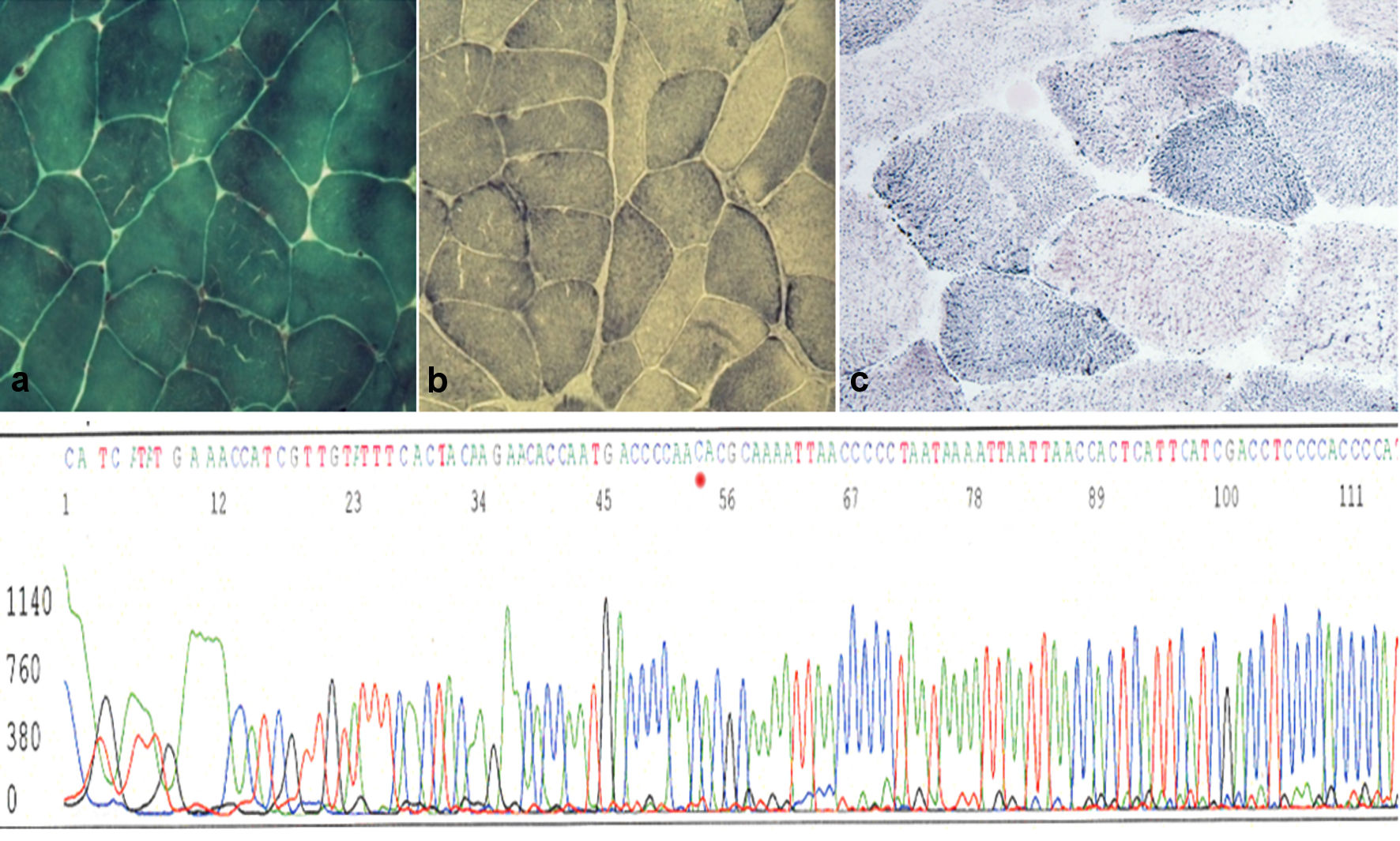 Click for large image | Figure 1. Upper panel: (a) muscle biopsy section stained for Gomori’s trichrome shows muscle fibers with normal distribution of mitochondria (magnification × 40). (b) Succinate dehydrogenase (SDH) staining shows a similar picture with some low dark blue fibers (magnification × 40) compared to a healthy control (c). Lower panel: automated sequence analysis of the region encompassing the m.14757T>C mutation of the proband’s mtDNA in muscle. |
Treatment
The patient benefited significantly from conventional treatment recommended for heart failure.
Follow-up and outcomes
Short-term follow-up after 8 weeks showed complete recovery of heart failure.
| Discussion | ▴Top |
DCM is not only due to nDNA gene mutations expressed in the myocardium [16], but also due to mtDNA mutations [17]. Accordingly, several studies found reduced activities of respiratory chain complexes in patients with DCM, where complex-III being the most frequently decreased [18-21]. Most frequently cytochrome-b gene mutations were shown to be responsible for DCM, as confirmed by the regularly updated database Mitomap [15]. Myocardial tissue from the index patient was not available for molecular genetic analysis, but screening of MT-CYB in skeletal muscle revealed a novel modification from the Cambridge mtDNA sequence (M4T) with slightly reduced complex-III activity. The mutation was absent in the control population and was not reported as neutral polymorphism despite sequencing of MT-CYB in 2,704 normal individuals from different ethnic backgrounds [15]. The physico-chemical properties of M and T residues are dissimilar. Methionine is medium hydrophobic and threonine is medium polar. The genetic code seems to have evolved towards minimising changes of the physico-chemical properties and to favor the rate of synonymous substitutions [22], suggesting that AA replacements resulting in a dissimilar AA are generally more deleterious than replacements resulting in an AA with similar properties.
We predicted the possible effect of the mutation on the structure and function of complex-III by using Polyphen [23]. As shown in Table 2, the substitution was predicted as “possibly damaging”, suggesting that codon-4 might be under evolutionary selective pressure and that the 4M variant is likely to be functional.
Click to view | Table 2. Prediction Data of Mutations Detected in the Index Patient |
The exact effects of the M4T mutation on the enzyme activity were difficult to elucidate. Previously reported missense mutations in MT-CYB impaired the assembly or stability of the complex, thus leading to either a dramatic decrease in the amount of assembled enzyme or to reduced catalytic activity of the complex with little effect on its assembly [24]. Some mutations in MT-CYB were investigated in the yeast system. In humans, the G34S mutation has been reported in a patient suffering from exercise intolerance [25] and the substitution of the corresponding glycine by aspartate led to extinction of the bc1 complex activity [26]. The G339E mutation manifested as myopathy and the same mutation abolished the bc1 complex assembly in yeast [27, 28]. The G251D mutation, which substitutes a glycine by aspartate at np251 of MT-CYB, causes histiocytoid cardiomyopathy. This mutation is located in the intermembrane space loop connecting the fifth and sixth transmembrane segments. Analyses of myocardial mitochondria revealed a defect of the succinate cytochrome-c-oxidoreductase activity and of the cytochrome-b assembly, documenting the importance of residue 251 for proper bc1 function [29, 30].
Limitations of the study were that the mechanism by which the mutation determined the phenotype could not be demonstrated and that the OXPHOS enzyme analysis was not carried out in myocardial tissue.
In conclusion, the variant M4T in MT-CYB was considered pathogenic because it was consistent with the biochemical complex-III defect, it was not found in 100 healthy controls, and because it has not been reported as neutral polymorphism despite extensive sequencing of MT-CYB in 2,704 healthy individuals from different ethnic backgrounds. Future studies of transgenic mice with defined mutations may help assessing their contribution to the pathophysiological background of DCM.
Acknowledgments
This work was supported by the Ministry of Higher Education, Scientific Research and Technology in Tunisia. We wish to thank the patients and their families for their support and collaboration.
Financial Disclosure
No funding was received.
Conflict of Interest
The authors declare no conflict of interest.
Informed Consent
Informed consent was obtained from the patient.
Author Contributions
SZ: design, literature search, discussion, first draft, and critical comments; JF, SM, FO, SBA, and RB: literature search, discussion, critical comments, and final approval.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
| References | ▴Top |
- Dec GW, Fuster V. Idiopathic dilated cardiomyopathy. N Engl J Med. 1994;331(23):1564-1575.
doi pubmed - Silvestri G, Bertini E, Servidei S, Rana M, Zachara E, Ricci E, Tonali P. Maternally inherited cardiomyopathy: a new phenotype associated with the A to G AT nt.3243 of mitochondrial DNA (MELAS mutation). Muscle Nerve. 1997;20(2):221-225.
doi - Takeda N. Cardiomyopathies and mitochondrial DNA mutations. Mol Cell Biochem. 1997;176(1-2):287-290.
doi pubmed - Vilarinho L, Santorelli FM, Rosas MJ, Tavares C, Melo-Pires M, DiMauro S. The mitochondrial A3243G mutation presenting as severe cardiomyopathy. J Med Genet. 1997;34(7):607-609.
doi pubmed - Barth E, Stammler G, Speiser B, Schaper J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol. 1992;24(7):669-681.
doi - Berry EA, Guergova-Kuras M, Huang LS, Crofts AR. Structure and function of cytochrome bc complexes. Annu Rev Biochem. 2000;69:1005-1075.
doi pubmed - Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290(5806):457-465.
doi pubmed - Marin-Garcia J, Goldenthal MJ, Moe GW. Mitochondrial pathology in cardiac failure. Cardiovasc Res. 2001;49(1):17-26.
doi - Marin-Garcia J, Goldenthal MJ, Moe GW. Abnormal cardiac and skeletal muscle mitochondrial function in pacing-induced cardiac failure. Cardiovasc Res. 2001;52(1):103-110.
doi - Moe GW, Marin-Garcia J, Konig A, Goldenthal M, Lu X, Feng Q. In vivo TNF-alpha inhibition ameliorates cardiac mitochondrial dysfunction, oxidative stress, and apoptosis in experimental heart failure. Am J Physiol Heart Circ Physiol. 2004;287(4):H1813-1820.
doi pubmed - Buchwald A, Till H, Unterberg C, Oberschmidt R, Figulla HR, Wiegand V. Alterations of the mitochondrial respiratory chain in human dilated cardiomyopathy. Eur Heart J. 1990;11(6):509-516.
doi pubmed - Jarreta D, Orus J, Barrientos A, Miro O, Roig E, Heras M, Moraes CT, et al. Mitochondrial function in heart muscle from patients with idiopathic dilated cardiomyopathy. Cardiovasc Res. 2000;45(4):860-865.
doi - Unverferth DV, Lee SW, Wallick ET. Human myocardial adenosine triphosphatase activities in health and heart failure. Am Heart J. 1988;115(1 Pt 1):139-146.
doi - Rustin P, Chretien D, Bourgeron T, Gerard B, Rotig A, Saudubray JM, Munnich A. Biochemical and molecular investigations in respiratory chain deficiencies. Clin Chim Acta. 1994;228(1):35-51.
doi - MITOMAP: a human mitochondrial genome database. http://www.mitomap.org.
- Fatkin D, Graham RM. Molecular mechanisms of inherited cardiomyopathies. Physiol Rev. 2002;82(4):945-980.
doi pubmed - Casademont J, Miro O. Electron transport chain defects in heart failure. Heart Fail Rev. 2002;7(2):131-139.
doi pubmed - Marin-Garcia J, Goldenthal MJ, Pierpont ME, Ananthakrishnan R. Impaired mitochondrial function in idiopathic dilated cardiomyopathy: biochemical and molecular analysis. J Card Fail. 1995;1(4):285-291.
doi - Rosca MG, Vazquez EJ, Kerner J, Parland W, Chandler MP, Stanley W, Sabbah HN, et al. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res. 2008;80(1):30-39.
doi pubmed - Sharov VG, Goussev A, Lesch M, Goldstein S, Sabbah HN. Abnormal mitochondrial function in myocardium of dogs with chronic heart failure. J Mol Cell Cardiol. 1998;30(9):1757-1762.
doi pubmed - Sharov VG, Todor AV, Silverman N, Goldstein S, Sabbah HN. Abnormal mitochondrial respiration in failed human myocardium. J Mol Cell Cardiol. 2000;32(12):2361-2367.
doi pubmed - Xia X, Li WH. What amino acid properties affect protein evolution? J Mol Evol. 1998;47(5):557-564.
doi pubmed - POLYPHEN: prediction of functional effect of human nsSNPs. http://www.genetics.bwh.harvard.edu.
- Fisher N, Meunier B. Effects of mutations in mitochondrial cytochrome b in yeast and man. Deficiency, compensation and disease. Eur J Biochem. 2001;268(5):1155-1162.
doi pubmed - Andreu AL, Hanna MG, Reichmann H, Bruno C, Penn AS, Tanji K, Pallotti F, et al. Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. N Engl J Med. 1999;341(14):1037-1044.
doi pubmed - Coppee JY, Brasseur G, Brivet-Chevillotte P, Colson AM. Non-native intragenic reversions selected from Saccharomyces cerevisiae cytochrome b-deficient mutants. Structural and functional features of the catalytic center N domain. J Biol Chem. 1994;269(6):4221-4226.
doi - Lemesle-Meunier D, Brivet-Chevillotte P, di Rago JP, Slonimski PP, Bruel C, Tron T, Forget N. Cytochrome b-deficient mutants of the ubiquinol-cytochrome c oxidoreductase in Saccharomyces cerevisiae. Consequence for the functional and structural characteristics of the complex. J Biol Chem. 1993;268(21):15626-15632.
doi - Andreu AL, Bruno C, Shanske S, Shtilbans A, Hirano M, Krishna S, Hayward L, et al. Missense mutation in the mtDNA cytochrome b gene in a patient with myopathy. Neurology. 1998;51(5):1444-1447.
doi pubmed - Papadimitriou A, Neustein HB, Dimauro S, Stanton R, Bresolin N. Histiocytoid cardiomyopathy of infancy: deficiency of reducible cytochrome b in heart mitochondria. Pediatr Res. 1984;18(10):1023-1028.
doi pubmed - Andreu AL, Checcarelli N, Iwata S, Shanske S, DiMauro S. A missense mutation in the mitochondrial cytochrome b gene in a revisited case with histiocytoid cardiomyopathy. Pediatr Res. 2000;48(3):311-314.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.