

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 13, Number 2, February 2022, pages 56-60

Glutaric Aciduria Type 2 Presenting in Adult Life With Hypoglycemia and Encephalopathic Hyperammonemia
Rute Sousa Martinsa, e ![]() , Arlindo Guimasa, b
, Arlindo Guimasa, b ![]() , Sara Rochaa, b
, Sara Rochaa, b ![]() , Rosa Ribeiroa, b
, Rosa Ribeiroa, b ![]() , Esmeralda Martinsb
, Esmeralda Martinsb ![]() , Manuela Almeidab, c, d
, Manuela Almeidab, c, d ![]() , Dulce Quelhasb, c, d
, Dulce Quelhasb, c, d ![]()
aDepartment of Internal Medicine, Centro Hospitalar Universitario do Porto, Porto, Portugal
bReference Centre for Inborn Errors of Metabolism, Centro Hospitalar Universitario do Porto, Porto, Portugal
cCentro de Genetica Medica Doutor Jacinto de Magalhaes, Centro Hospitalar e Universitario do Porto, Porto, Portugal
dUnit for Multidisciplinary Research in Biomedicine, Abel Salazar Institute of Biomedical Sciences, University of Porto, Porto, Portugal
eCorresponding Author: Rute Sousa Martins, Department of Internal Medicine, Centro Hospitalar Universitario do Porto, Largo Prof. Abel Salazar 4099-001 Porto, Portugal
Manuscript submitted November 3, 2021, accepted February 4, 2022, published online February 16, 2022
Short title: Glutaric Aciduria Type 2 in Adult Life
doi: https://doi.org/10.14740/jmc3840
| Abstract | ▴Top |
Glutaric aciduria type 2 is a rare inborn disease of fatty acid metabolism. The clinical manifestation of this disease is heterogeneous and ranges from severe neonatal forms to mild late-onset forms. We present a case of a previously healthy 22-year-old woman with unexplainable hypoglycemia and encephalopathic hyperammonemia. Acylcarnitine profile and organic acids analysis were compatible with glutaric aciduria type 2. On suspicion of this disease, the patient started supplements with carnitine and riboflavin, along with hemodialysis with a complete recovery. The genetic test confirmed the diagnosis. Glutaric aciduria type 2 has no cure and the metabolic decompensation can be a severe event, but treatable and preventable, if this pathology gets recognized.
Keywords: Glutaric aciduria type 2; Rare disease; Hypoglycemia
| Introduction | ▴Top |
Glutaric aciduria type 2, or multiple acyl-CoA dehydrogenase deficiency, is an autosomal recessive inherited disorder of the mitochondrial electron transfer flavoprotein chain causing dysfunction in fatty acid, organic acid and choline metabolism [1, 2].
It is caused mostly by homozygous or compound heterozygous mutations that lead to deficiency in electron-transferring-flavoprotein, alpha and beta polypeptide (ETFA and ETFB), and electron-transferring-flavoprotein dehydrogenase (ETFDH), causing defects in the mitochondrial transport of electrons from the acyl-CoA dehydrogenases (short-, medium-, and long-chain) to ubiquinone (CoQ10) [3] of the mitochondrial electron transport chain. Consequently, there is accumulation of different substrates to those enzymes.
It is a clinically heterogeneous disease with three clinical forms: a neonatal form with congenital anomalies, a neonatal-onset form without congenital anomalies, and a late-onset form. Neonatal forms are severe, typically present with hypo- or non-ketotic hypoglycemia and metabolic acidosis during the first days of life [4, 5]. This type of presentation is typically fatal. Later-onset glutaric aciduria type 2 may occur in the first few months of life, or later in childhood or adulthood, with intermittent episodes of hypoglycemia, vomiting or metabolic acidosis. Abnormalities are related to fatty infiltrations of the muscle (presenting as proximal myopathy), liver (with hepatomegaly, steatosis and cirrhosis [2]), myocardium (cardiomyopathy), and kidneys, because these tissues use fatty acids as a primary source of energy [4, 5]. In addition, laboratory findings may include hyperammonemia and elevated liver transaminases [6].
Treatment aims to prevent metabolic decompensation through low-protein and low-fat diets with oral supplements of carnitine and riboflavin, while hypoglycemic attacks are managed with oral or intravenous glucose.
We present a case of late-onset glutaric aciduria type 2 characterized by hypoglycemia and encephalopathy in a previously healthy patient.
| Case Report | ▴Top |
Investigations
We present a case of a healthy 22-year-old female, with no consanguinity in her family, who came to the emergency room with anorexia, vomiting and nausea in the past month associated with a significant reduction of food intake and weight loss of at least 10 kg (15% of her body weight), and no fever was reported. In admission, the patient was conscious but confused with a Glasgow Coma Scale of 14 points (E4V4M6) and, on the physical examination, she had Kussmaul breathing pattern and a fruity breath odour. The patient had metabolic acidosis with an elevated anion gap, a normal lactate level, and a hypoglycemia of 55 mg/dL. No alterations were seen on the hepatic or renal functions, and the ion levels were within normal, but there was an abundant presence of ketone bodies in her urine sample. The serum salicylate level was negative, and the levels of cortisol, insulin and thyroid function were normal.
Diagnosis
The patient presented a worsening of her neurological status with a Glasgow Coma Scale of 10 points (E2V4M4), despite the improvement of her glucose levels. The cerebral computed tomography (CT) scan was normal and no lumbar puncture was performed. She was then admitted in level 2 intensive care. She had no signs of infection, alcohol, or drug abuse intoxications, which could explain the encephalopathy. Because she had metabolic acidosis with elevated anion gap, a low blood-glucose level and presence of ketone bodies, a deficiency in energetic metabolism was suspected and a metabolic study was performed. The patient’s ammonia levels were elevated (261 µmol/L), without any hepatic disorder or acute dysfunction.
Treatment
She started immediately with glucose 10% fluids, carnitine, riboflavin, and sodium benzoate, due to the suspected energetic metabolic deficiency. In addition to the drug therapy, continuous venovenous hemodiafiltration (CVVHDF) was performed for 3 days, to reduce the level of ammonia and accumulated acids, with a drastic neurologic improvement.
An acylcarnitine analysis was performed and showed a low level of free carnitine and elevated levels of C14 and C16:1 acylcarnitine, while her urine analysis showed excretion of multiple organic acids compatible with glutaric aciduria type 2. The amino acid profile showed elevated proline, lysine and tyrosine with no alteration of glutamine, citrulline or arginine, suggesting that a primary urea cycle disorder was of low probability.
Follow-up and outcomes
The patient recovered her baseline mental status, and the laboratory parameters improved with the normalization of hypoglycemia, hyperammonemia and metabolic acidosis. She was discharged after 1 week with a diet restriction of proteins and lipids along with oral supplements of carnitine, sodium benzoate and riboflavin.
A later diagnosis of glutaric aciduria type 2 was confirmed with gene analysis where two pathogenic mutations were found in ETFDH gene: c.245C>T(p.S82F) and c.1601C>T(p.P534L).
Since the diagnosis, the patient is closely monitored at our reference center of inherited metabolic diseases and maintains an active life with oral supplements of carnitine and riboflavin, along with restricted diet proteins and lipids, and a higher carbohydrates intake. Figures 1 and 2 show the patient acylcarnitine profile in the beginning of the diagnosis and its evolution with the treatment described above. The hyperammonemia was resolved and the sodium benzoate was removed from ambulatory therapy.
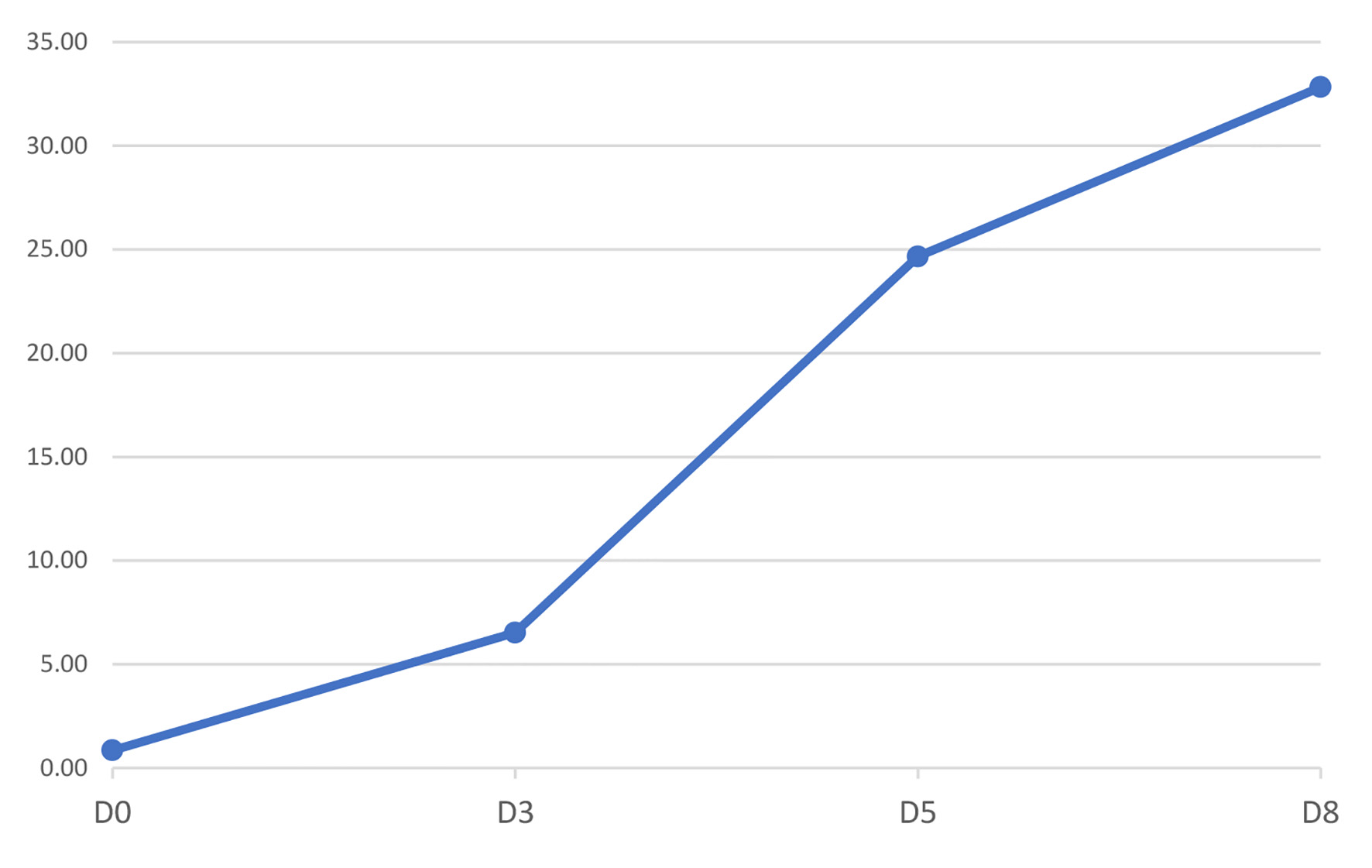 Click for large image | Figure 1. Figure 1. Evolution of free carnitine (C0) profile since diagnosis (D0) until discharge (D8). Units are in µM. |
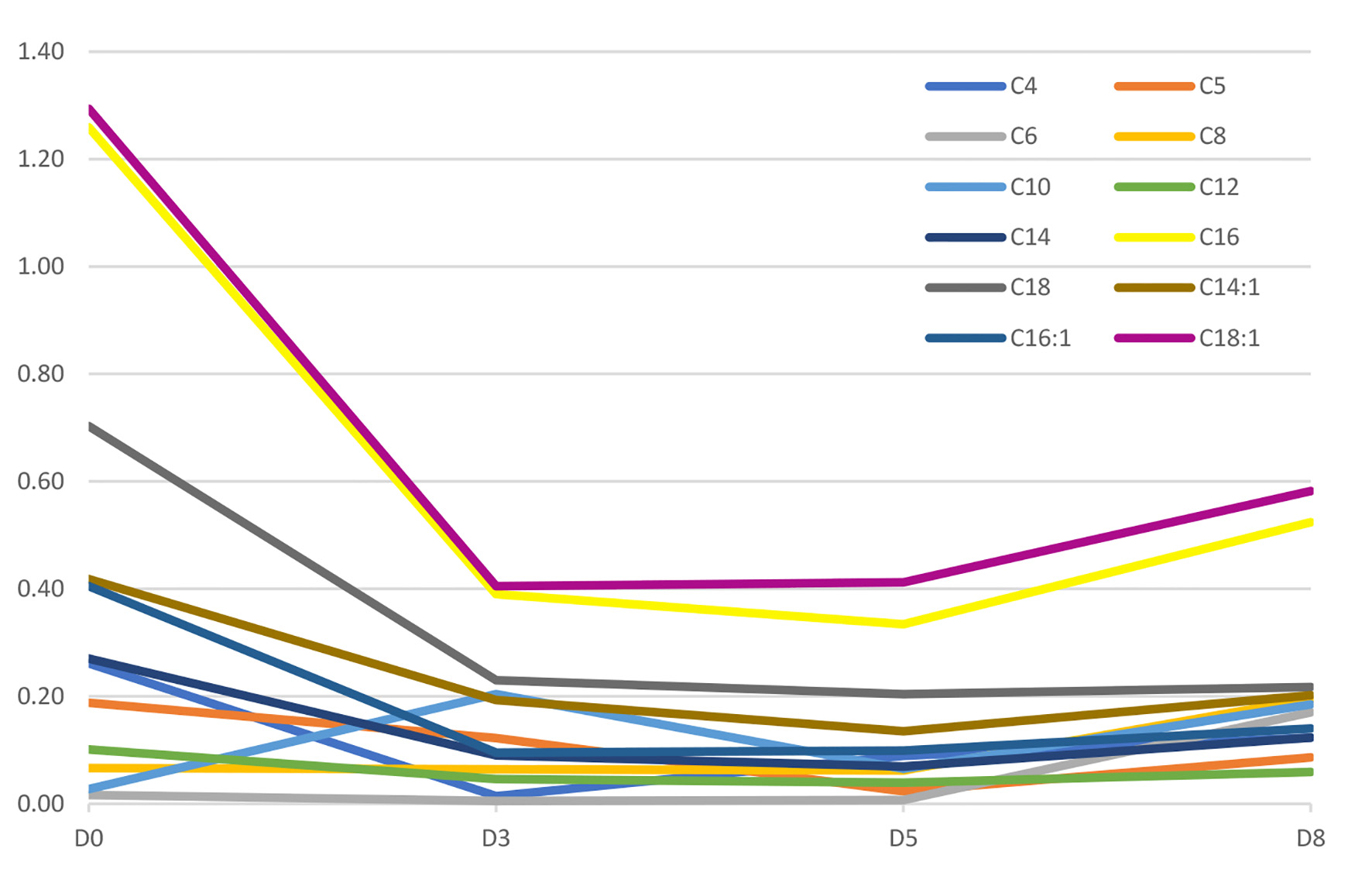 Click for large image | Figure 2. Figure 2. Evolution of acylcarnitine profile since diagnosis (D0) until discharge (D8). Units are in µM. |
| Discussion | ▴Top |
We report a case of late-onset glutaric aciduria type 2 with the first acute decompensation in adult life. The acute crisis has several bizarre metabolic findings that could be challenging for the diagnosis like hypoglycemia, metabolic acidemia with elevated anion gap, and hyperammonemic encephalopathy because they are non-specific.
A high suspicion index is mandatory to perform the essential metabolic study as soon as possible to guide the clinician to the adequate diagnosis. The metabolic team of our center was involved at an early stage in the treatment of this patient, having a key contribution for the diagnosis and resolution of the decompensation.
The plasmatic aminogram is central for the understanding of the hyperammonemia and to evaluate the nitrogen balance. In this patient, the typical finding of urea cycle disorders was not found, leading to a study for a secondary cause of hyperammonemia.
The measurement of organic acid in the urine shows an acid accumulation in the blood, which is then eliminated by the kidney. Urine organic acid analysis in glutaric aciduria type 2 reveals an elevated excretion of lactic and glutaric acids, along with a large number of other hydroxy and dicarboxylic acids [3]. It is important to state that stable forms of glutaric aciduria type 2 can have normal levels of urine organic acids with elevated levels only in symptomatic periods. In this patient, the excretion of organic acid in the urine persisted after this event.
The acylcarnitine profile is a very useful diagnostic tool that shows the accumulation of acyl skeletons of lipids and other metabolites that bind to carnitine; this patient shows the typically elevated C4 to C18-acylcarnitines [7].
The sweetish or characteristic fruity odour of patients with glutaric aciduria type 2 is due to the accumulation of isovaleric acid and its metabolites, and can also be found in patients with isovaleric aciduria [8]. This simple finding in physical examination can help the clinicians direct their study into the metabolic etiology behind the case presented.
Given the rarity of the situation, and since delaying the beginning of the treatment can have deleterious consequences most of the time, it is more important to identify the group to which the disease belongs to and start, if possible, the adequate treatment to reverse the metabolic disarrangement. In this case, the glucose, riboflavin and dialysis were crucial to the treatment of this patient.
Riboflavin is a precursor of the essential flavin adenine dinucleotide, cofactor for the deficient enzymes, and riboflavin deficiency may mimic glutaric aciduria type 2. Riboflavin-responsive mutations have also been described, most frequently in ETFDH gene [9]. An early intake of this vitamin may be crucial to potentially reverse the enzymatic disfunction observed in this disease.
Depletion of body stores of carnitine even with mild disease is expected [3, 10], so carnitine supplement is necessary, but usually not sufficient to treat the condition. In our patient, a significant improvement was observed after initiating high doses of carnitine (Figs. 1 and 2).
Secondary hyperammonemia (due to the combined effects of substrate depletion and urea cycle inhibition by toxic acylcarnitine species) [6] was treated with CVVHDF and sodium benzoate. There are no established guidelines on which ammonia value justifies initiating extracorporeal dialysis technique and the best timing for initiation in these patients is yet to be defined. However, it has been proposed to start when blood ammonia levels are higher than 250 µmol/L and in the presence of severe encephalopathy [11]. Both continuous and intermittent techniques are effective in removing ammonia, differing only in the removal rate [11, 12]. However, the choice of the technique depends on the availability and experience of each center and on which is possible to start earlier.
In our patient, CVVHDF was performed because of the degradation of the mental status and due to the unpredictability of hyperammonemia’s evolution in the disorder.
Hypoglycemia can be explained since the impaired fat oxidation, and consequent decrease of hepatic gluconeogenesis, leads to greater dependence upon carbohydrate oxidation [2, 10]. The energetic failure of hepatocyte could explain the inability to maintain normoglycemia and abnormal ammonia clearance.
Learning points
The presence of hypoglycemia, metabolic acidosis and hyperammonemic encephalopathy, as they are all signs and symptoms of metabolic disarrangement, should prompt the study to an inborn error of metabolism, especially after excluding the most common etiologies for these frequent findings. Remembering that these rare diseases can appear even in adult life is crucial for a quick diagnosis and treatment.
Conclusion
The inherited metabolic errors of metabolism are rare and glutaric aciduria type 2 has a low prevalence at birth 1:250,000 [13], thus presenting a very difficult diagnosis. There are some clues that must be a red flag for clinicians, and we believe that this case is a good example of that, having a constellation of several of those that triggered a proper investigation and treatment.
This case serves as a reminder that late-onset inherited metabolic diseases like glutaric aciduria type 2 may manifest in adult life and those acute events could be severe and life-threatening, but the correct diagnosis and rapid treatment is extremely important to prevent lesions and death.
The communication with the metabolic team of any reference center is crucial to help define the correct diagnostic approach and treatment, when possible.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Informed consent has been obtained from the patient.
Author Contributions
Rute Sousa Martins was responsible for the conception and the writing of the manuscript. Arlindo Guimas and Dulce Quelhas contributed to the diagnostic investigation, data analysis, and the writing of the article. They also belong to the inborn errors of the metabolism team. Sara Rocha, Rosa Ribeiro, Esmeralda Martins and Manuela Almeida belong to the inborn errors of the metabolism team and assisted in revisions of final drafts. All authors approved the final manuscript and are accountable for all aspects of the work.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Wasant P, Kuptanon C, Vattanavicharn N, Liammongkolkul S, Ratanarak P, Sangruchi T, Yamaguchi S. Glutaric aciduria type 2, late onset type in Thai siblings with myopathy. Pediatr Neurol. 2010;43(4):279-282.
doi pubmed - Soldath P, Lund A, Vissing J. Late-onset MADD: a rare cause of cirrhosis and acute liver failure? Acta Myol. 2020;39(1):19-23.
- Nyhan WL, Hoffmann GF, Al-Aqeel AI, Barshop BA. Atlas of inherited metabolic diseases. Boca Raton, CRC Press. 2020.
doi - Angle B, Burton BK. Risk of sudden death and acute life-threatening events in patients with glutaric acidemia type II. Mol Genet Metab. 2008;93(1):36-39.
doi pubmed - al-Essa MA, Rashed MS, Bakheet SM, Patay ZJ, Ozand PT. Glutaric aciduria type II: observations in seven patients with neonatal- and late-onset disease. J Perinatol. 2000;20(2):120-128.
doi pubmed - Guerrero RB, Salazar D, Tanpaiboon P. Laboratory diagnostic approaches in metabolic disorders. Ann Transl Med. 2018;6(24):470.
doi pubmed - Vreken P, van Lint AE, Bootsma AH, Overmars H, Wanders RJ, van Gennip AH. Rapid diagnosis of organic acidemias and fatty-acid oxidation defects by quantitative electrospray tandem-MS acyl-carnitine analysis in plasma. Adv Exp Med Biol. 1999;466:327-337.
doi pubmed - Gordon N. Glutaric aciduria types I and II. Brain Dev. 2006;28(3):136-140.
doi pubmed - Wen B, Dai T, Li W, Zhao Y, Liu S, Zhang C, Li H, et al. Riboflavin-responsive lipid-storage myopathy caused by ETFDH gene mutations. J Neurol Neurosurg Psychiatry. 2010;81(2):231-236.
doi pubmed - Dusheiko G, Kew MC, Joffe BI, Lewin JR, Mantagos S, Tanaka K. Recurrent hypoglycemia associated with glutaric aciduria type II in an adult. N Engl J Med. 1979;301(26):1405-1409.
doi pubmed - Haberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, Karall D, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32.
doi pubmed - Slack AJ, Auzinger G, Willars C, Dew T, Musto R, Corsilli D, Sherwood R, et al. Ammonia clearance with haemofiltration in adults with liver disease. Liver Int. 2014;34(1):42-48.
doi pubmed - Schulze A, Lindner M, Kohlmuller D, Olgemoller K, Mayatepek E, Hoffmann GF. Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications. Pediatrics. 2003;111(6 Pt 1):1399-1406.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.