

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 13, Number 4, April 2022, pages 145-150

An Immunoglobulin A Vasculitis Case Without Skin Symptoms Complicated With Severe Abdominal Symptoms
Tomonobu Satoa, b ![]() , Yasuyoshi Hiramatsua, Hisato Segoea, Kota Watanabea, Haruki Shiraishia, Yuji Maruoa, Norio Satoa, Takashi Suganumaa, Makoto Mikawaa
, Yasuyoshi Hiramatsua, Hisato Segoea, Kota Watanabea, Haruki Shiraishia, Yuji Maruoa, Norio Satoa, Takashi Suganumaa, Makoto Mikawaa
aDepartment of Pediatrics, Kitami Red Cross Hospital, Kitami 090-8666, Japan
bCorresponding Author: Tomonobu Sato, Department of Pediatrics, Kitami Red Cross Hospital, Kitami 090-8666, Japan
Manuscript submitted December 29, 2021, accepted February 11, 2022, published online March 25, 2022
Short title: IgAV With Lacking Skin Symptoms
doi: https://doi.org/10.14740/jmc3893
| Abstract | ▴Top |
Immunoglobulin A vasculitis (IgAV) primarily affects childhood and can be categorized as immune complex vasculitis. It typically presents with purpura, abdominal pain, arthritis, and nephritis. IgAV can be diagnosed without hesitation when the characteristic skin lesions appear at onset; however, in cases where the abdominal symptoms precede the skin rash or there is no purpura at all, diagnosis can be challenging. Delayed diagnosis of IgAV may be associated with serious abdominal complications, such as gastrointestinal perforation. Here, we describe a girl with IgAV complicated with severe abdominal symptoms and lacking purpura. Despite this lack, the patient’s elevated levels of D-dimer and C-reactive protein (CRP), suggestive of vasculitis, and localized small bowel intestinal wall thickening suggested IgAV. After administration of steroids relieved the abdominal symptoms and hypoalbuminemia, treatment was discontinued. Given the limited reports of patients with IgAV complicated with severe abdominal symptoms and no skin symptoms, the diagnosis and management process remains unclear. Therefore, it is imperative to consider IgAV as a differential diagnosis in patients with severe abdominal symptoms. Furthermore, we suggest checking D-dimer, CRP, and coagulation factor XIII activity levels in these patients.
Keywords: Immunoglobulin A vasculitis; Child; Abdominal symptoms
| Introduction | ▴Top |
Immunoglobulin A vasculitis (IgAV), previously known as Henoch-Schonlein purpura, is an immune complex vasculitis mainly caused by inflammation of small vessels, according to the 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides [1]. IgAV largely affects children and can evoke purpura, arthralgia, nephritis, and acute abdominal pain. The diagnosis of IgAV is most clear when the characteristic purpura appears at onset; however, in cases where abdominal symptoms precede the skin rash or when skin symptoms are absent, diagnosis can be difficult. Delayed diagnosis of IgAV can be associated with serious complications, such as gastroduodenal ulcers and gastrointestinal perforation.
Here, we report the case of a 3-year-old girl with IgAV who demonstrated recurrent abdominal pain but lacked skin symptoms.
| Case Report | ▴Top |
Investigations
A 3-year-old girl developed loss of appetite, vomiting, and intermittent periumbilical abdominal pain lasting 5 days prior to visiting our hospital. She demonstrated no obvious fever or bloody stool; however, her serum C-reactive protein (CRP) level was mildly elevated. She was temporarily admitted to our ward with a diagnosis of acute gastroenteritis, and discharged from our hospital after her symptoms improved with fluid replacement. However, the recurrence of intermittent abdominal pain appearing 1 day following discharge resulted in re-admission. She had no significant prior medical history other than abdominal pain and vomiting, and had been diagnosed with bacterial enteritis 8 months before admission. On admission, her body temperature was 37.2 °C, she had a mildly distended abdomen, and she appeared generally unwell. She had no overt skin symptoms such as palpable purpura, and no aphthous stomatitis or perianal lesions were observed. This patient had no family history of inflammatory bowel disease (IBD).
Diagnosis
Laboratory findings are presented in Table 1. The peripheral white blood cell counts and platelets were 24,930/µL and 64.7 × 104/µL, respectively. Blood chemistry tests revealed increased levels of CRP and hypoproteinemia without proteinuria. Although the findings of the coagulation system were within the normal range, increased levels of D-dimer, indicating vasculitis, were observed. Other laboratory tests were unremarkable, including serum immunoglobulin, autoantibodies, and cytomegalovirus antibody titers. The fecal occult blood test result was positive. Abdominal radiography revealed a gasless abdomen, and abdominal ultrasonography (US) and contrast-enhanced abdominal computed tomography (CT) both revealed significant thickening of the small bowel walls (Fig. 1). No findings suggestive of intussusception were found.
 Click to view | Table 1. Laboratory Findings on Admission |
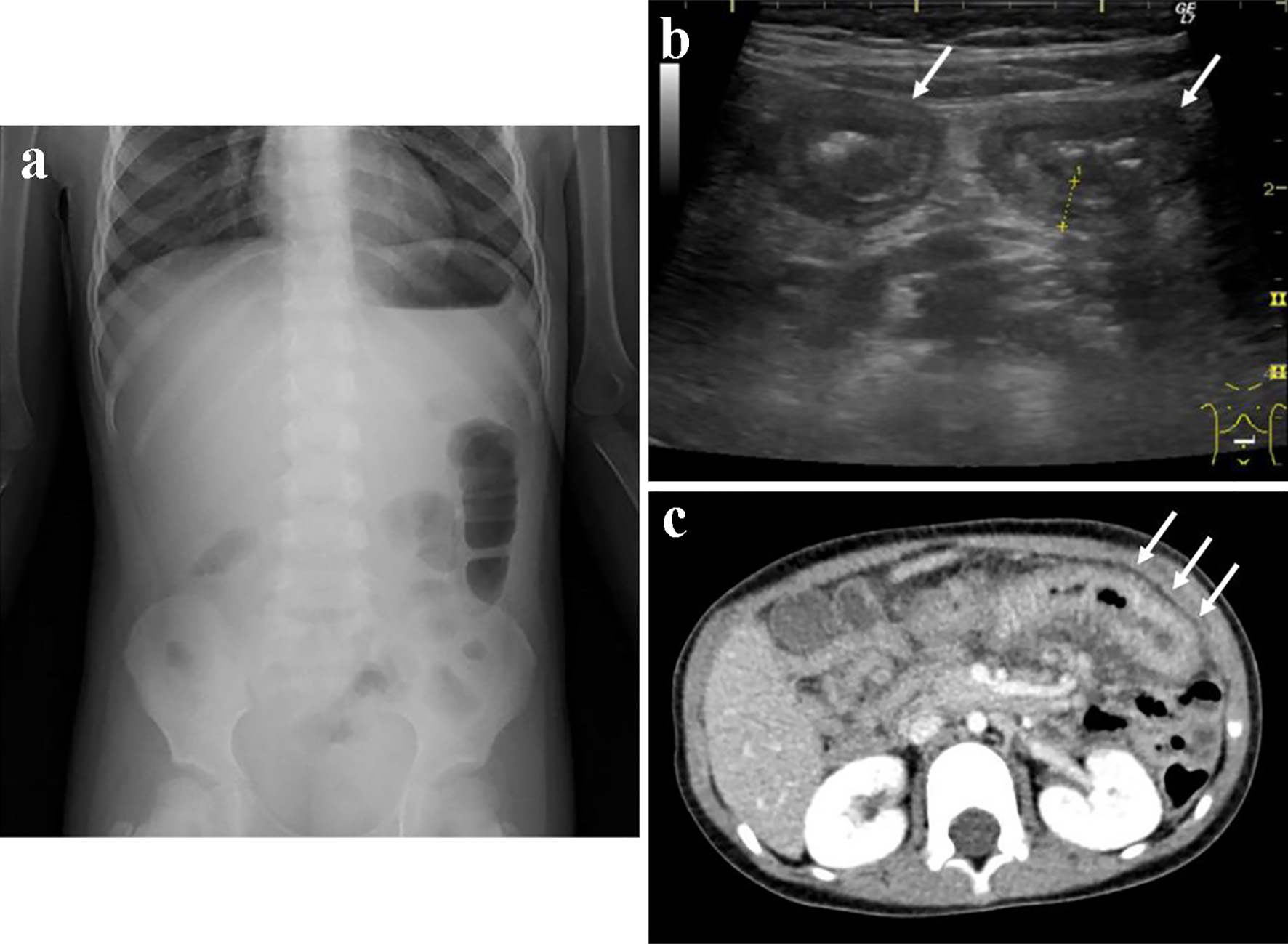 Click for large image | Figure 1. Findings on an abdominal roentgenography, an ultrasonography, and a contrast computed tomography at disease onset. At disease onset, roentgenography revealed a gasless abdomen and an image of a partially enlarged colon (a). Abdominal ultrasonography and enhanced computed tomography showed images of the thickened wall of the small intestine (b, c, white arrow). |
After screening for the cause of her abdominal symptoms, it was initially determined that bacterial enteritis was the cause of her symptoms; therefore, follow-up was performed with oral antibiotics, fosfomycin, and fluid replacement (Fig. 2). However, antibiotic therapy did not improve her abdominal symptoms. Despite the lack of characteristic purpura and arthralgia, findings such as poor therapeutic effects with antibiotics, elevated D-dimer levels suggestive of vasculitis on blood tests, and localized small bowel intestinal wall thickening suggested that the patient was suffering from IgAV. Furthermore, she demonstrated progressive abdominal distension and systemic edema; subsequent levels of serum total protein and serum albumin were 4.8 and 2.4 g/dL, respectively, indicating the deterioration of hypoproteinemia. No proteinuria was observed. She was administered intravenous albumin, and her edema improved temporarily. Although evaluation using protein-leakage scintigraphy and measurement of α1-antitrypsin concentration in stool was not performed, the possibility of a protein-losing enteropathy (PLE) complication was tentatively considered.
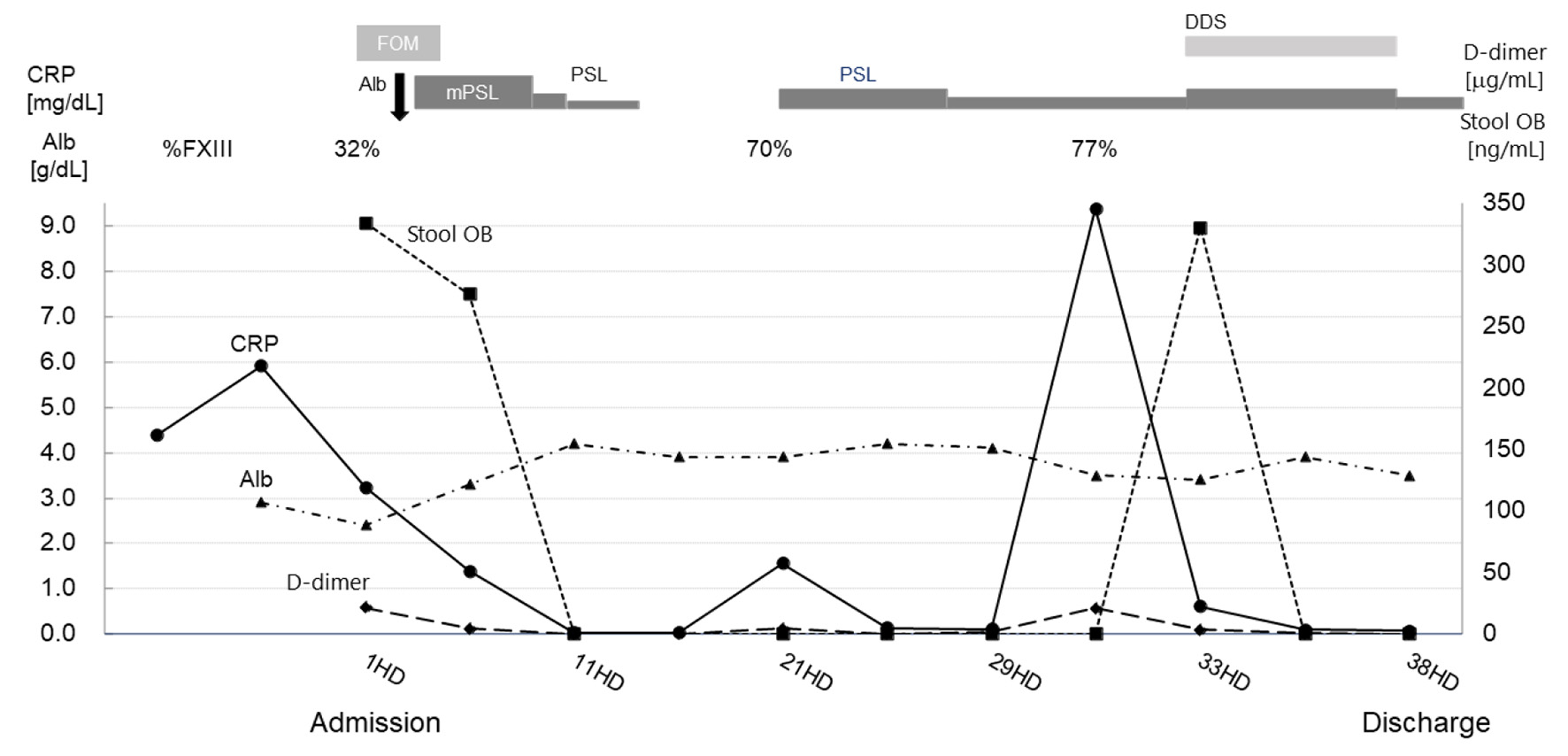 Click for large image | Figure 2. Clinical course in relation to laboratory findings. The horizontal axis shows days after the patient’s admission, and the left vertical axis shows the patient’s serum levels of CRP and Alb. The right vertical axis shows the levels of D-dimer and the quantitative value of stool OB. The upper row shows the transition of FXIII activity and treatment content. The polygonal lines show the transition of the inspection values. CRP: C-reactive protein; Alb: serum albumin; %FXIII: coagulation factor XIII activity; stool OB: stool occult blood; FOM: fosfomycin; PSL: prednisolone; mPSL: methylprednisolone; DDS: diaminodiphenyl sulfone; HD: hospital days. |
Treatment
She was administered intravenous methylprednisolone (mPSL, 2 mg/kg/day), and her abdominal pain and fecal occult blood immediately improved. In addition, blood chemistry tests showed decreased CRP levels and increased serum albumin levels, and the abdominal distension and systemic edema resolved. Three days after admission, we confirmed that coagulation factor XIII (FXIII) activity was decreased to 32% (normal reference range: ≥ 70%), suggestive of IgAV. Her treatment was changed from intravenous mPSL to oral prednisolone (PSL), which was subsequently tapered and discontinued. However, tapering or discontinuation of PSL treatment led to returned abdominal pain accompanied by elevation of D-dimer and CRP levels. Her abdominal symptoms became dependent on PSL therapy, and diaminodiphenyl sulfone (DDS) was temporarily administered; however, this was discontinued due to liver dysfunction. On hospital day 38, her abdominal symptoms improved with PSL treatment, and she was discharged. At the outpatient ward, PSL was tapered over 2 weeks, and the PSL was withdrawn. Three days after the end of PSL treatment, she again presented with abdominal pain and bloody stools. She was referred to a pediatric gastrointestinal disease center hospital to rule out the presence of IBD, such as Crohn’s disease, by endoscopy. Esophagogastroduodenoscopy and colonoscopy showed only mild swelling of the ileocecal valve, and histopathology of the intestine revealed nonspecific inflammation. Although video capsule endoscopy showed a small amount of blood adhesion to the wall of the small intestine, no longitudinal ulcers or cobble stone appearance that would be suggestive of Crohn’s disease were observed. In addition, no findings suggestive of Meckel’s diverticulum were confirmed by scintigraphy. As a result, a diagnosis of IgAV was given based on the above screening tests and her clinical course.
Follow-up and outcomes
The gastrointestinal symptoms subsequently improved without PSL by means of strict diet control including intestinal rest and fat-restriction. Currently, 3 years have passed since discharge with no relapse of abdominal symptoms or onset of nephropathy observed.
| Discussion | ▴Top |
In this report, we describe a girl with IgAV complicated with severe abdominal symptoms that fully lacked skin lesions. It has been reported that IgAV has an annual incidence of 3 - 27 cases per 100,000 children [2, 3], with a peak age of onset between 4 and 6 years old and a higher prevalence in the Asian population [4, 5]. There are still many unclear points regarding the cause and pathophysiology of IgAV. The IgA immune complex is produced by reactions to exogenous substances such as bacteria (group A streptococcal infections in particular), viral infections, chemical agents, food, insect bites, vaccines, and deposits on small vessel walls [6, 7]. These substances activate the complement system by deposition of IgA immune complex to vessel walls, resulting in the production of cytokines and causing IgAV symptoms [8]. For example, the deposition of IgA immune complexes on the walls of blood vessels is thought to cause hyperpermeability of capillaries [9]. Gastrointestinal lesions in IgAV patients can occur anywhere; however, they occur most often in the proximal intestine, specifically the duodenum and small bowel, and gastrointestinal perforation and intussusception can occur in severe cases [10]. In this case, the edema of the intestinal walls localized to the small intestine detected by US and CT was one of the clues for diagnosis of IgAV.
To date, there have been limited reports of IgAV cases in which abdominal symptoms precede cutaneous symptoms, as well as cases in which purpura is completely absent. In approximately 10-36% of IgAV cases, abdominal symptoms precede purpura [11, 12]. Therefore, IgAV with absence skin symptoms requires a definitive diagnosis by gastrointestinal endoscopy or renal biopsy. Reports have also advocated the possibility that a condition called IgA enteropathy may exist in IgAV cases in which IgA deposits are observed on the walls of small blood vessels, and skin symptoms are lacking altogether [12, 13]. Regarding the diagnostic criteria for IgAV, the American College of Rheumatology criteria consider purpura to be one symptom in IgA cases; however, it is not mandatory for diagnosis [14]. Conversely, the European League Against Rheumatism/Pediatric Rheumatology European Society (EULAR/PReS) criteria rely on the mandatory appearance of purpura and one or more of the other characteristic clinical findings such as abdominal pain, arthralgia or arthritis, and any biopsy with IgA deposition to diagnose IgAV [15]. Thus, according to the EULAR/PReS diagnostic criteria, our case could not be strictly diagnosed with IgAV. The Rumpel-Leede test is known to be one of the means for confirming the presence of skin lesions in IgAV cases; however, because the positive rate of the test is low, IgAV cannot be ruled out even if the result is negative.
It is known that in IgAV cases that completely lack skin symptoms such as ours, a significant increase in the levels of D-dimer or fibrin/fibrinogen degradation products and a decrease in coagulation FXIII activity are observed as auxiliary diagnostic findings [16]. Therefore, when encountering purpura-free patients with severe abdominal symptoms, elevated D-dimer levels, and thickening of the intestinal wall with main lesions in the duodenum and small intestine, IgAV should be considered as a differential diagnosis, and treatment for IgAV should be started while waiting for the results of a coagulation FXIII activity test. In addition, the severity score of abdominal symptoms in IgAV cases proposed by Nagamori et al could also be useful [17]. Scores from six endpoints, such as white blood cell count and neutrophil count, levels of D-dimer, activity of coagulation FXIII, serum sodium concentration, and serum albumin levels can predict whether steroid therapy should be administered to patients with abdominal symptoms. In our case, the patient’s score was 9 out of 10 points, which was considered to be a severe case requiring steroid treatment. However, some cases where it is difficult to distinguish between IgAV and IBD, such as Crohn’s disease, might require endoscopic evaluation [10]. In IgAV cases, we need to confirm lesions of the upper gastrointestinal tract in the duodenum or small intestine. Endoscopic examination is generally difficult for children, and the risks of adverse effects such as intestinal perforation require careful judgment of its indications. Recently, there have been some reports on the usefulness of video capsule endoscopy (VCE) for gastrointestinal disorders in children [18]. The effectiveness of VCE in IgAV patients needs to be analyzed by further case study. In our case, VCE examination showed slight bleeding in the small intestine, and no recurrence of gastrointestinal symptoms was observed after discontinuation of PSL, which was a factor in denying her diagnosis of IBD.
We could not make a definitive diagnosis due to the lack of various tests necessary for the diagnosis, but it was suggested that she may have developed PLE. In our case, there was a decrease in serum albumin levels without abnormal urinary findings. In IgAV cases in which solely abdominal pain is the initial symptom or those cases in which purpura is completely absent, the risk of complications with hypoproteinemia is expected to increase because of the delayed diagnosis of IgAV. In IgAV patients, serum albumin, as well as other serum proteins, such as immunoglobulins and coagulation factors, may decrease; therefore, it is necessary to pay close attention to serious infections and bleeding tendency. Hence, IgAV patients need to be diagnosed as soon as possible, and appropriate treatment such as administration of PSL and replacement of deficient proteins need to be administered promptly.
In conclusion, we encountered an IgAV case that was difficult to diagnose because abdominal symptoms presented solely and without skin lesions. She demonstrated hypoalbuminemia and required albumin replacement. She was also steroid-dependent and had repeated diffuse abdominal pain due to tapering and discontinuation of PSL. In patients with unexplained abdominal pain, IgAV should be included as a differential diagnosis even in the absence of skin lesions, and IgAV should be suspected as soon as possible by referring to parameters such as the value of D-dimer and coagulation FXIII activity. In IgAV patients, hypogammaglobulinemia and decreased levels of coagulation factors could cause serious infectious diseases and bleeding; hence, early treatment with PSL or replacement therapy is required.
Learning points
In this case, we found an IgAV case that was difficult to diagnose because of predominant severe abdominal symptoms without skin lesions. Our case report highlights the importance of checking D-dimer, CRP, and coagulation factor XIII activity levels in these patients.
Acknowledgments
We would like to thank Dr. Shinichi Fujiwara, Department of Pediatrics, Sapporo Kosei General Hospital, for coordinating the endoscopic examination and further treatment of the patients.
Financial Disclosure
The authors declare that there is no funding for the publication of this article.
Conflict of Interest
The authors indicated no potential conflict of interest.
Informed Consent
The patient’s guardians signed an informed consent to publish this report.
Author Contributions
TS wrote the paper; YH, HS, KW, HS, YM, NS, TS and MM reviewed the paper and gave conceptual advice; all authors read and approved the final manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
DDS: diaminodiphenyl sulfone; EULAR/PReS: the European League Against Rheumatism/Pediatric Rheumatology European Society; FXIII: coagulation factor XIII; IBD: inflammatory bowel disease; IgAV: immunoglobulin A vasculitis; mPSL: methylprednisolone; PLE: protein-losing enteropathy; PSL: prednisolone; VCE: video capsule endoscopy
| References | ▴Top |
- Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, Flores-Suarez LF, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1-11.
doi pubmed - Ruperto N, Ozen S, Pistorio A, Dolezalova P, Brogan P, Cabral DA, Cuttica R, et al. EULAR/PRINTO/PRES criteria for Henoch-Schonlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part I: Overall methodology and clinical characterisation. Ann Rheum Dis. 2010;69(5):790-797.
doi pubmed - Piram M, Maldini C, Biscardi S, De Suremain N, Orzechowski C, Georget E, Regnard D, et al. Incidence of IgA vasculitis in children estimated by four-source capture-recapture analysis: a population-based study. Rheumatology (Oxford). 2017;56(8):1358-1366.
doi pubmed - Oni L, Sampath S. Childhood IgA Vasculitis (Henoch Schonlein Purpura)-Advances and Knowledge Gaps. Front Pediatr. 2019;7:257.
doi pubmed - Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schonlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet. 2002;360(9341):1197-1202.
doi - Trapani S, Micheli A, Grisolia F, Resti M, Chiappini E, Falcini F, De Martino M. Henoch Schonlein purpura in childhood: epidemiological and clinical analysis of 150 cases over a 5-year period and review of literature. Semin Arthritis Rheum. 2005;35(3):143-153.
doi pubmed - Jauhola O, Ronkainen J, Koskimies O, Ala-Houhala M, Arikoski P, Holtta T, Jahnukainen T, et al. Clinical course of extrarenal symptoms in Henoch-Schonlein purpura: a 6-month prospective study. Arch Dis Child. 2010;95(11):871-876.
doi pubmed - Stacy PA, Edward F. Henoch-Schonlein Purpura. Nelson Textbook of Pediatrics. Vol. 19th. Elsevier, Philadelphia. 2011; 868-871.
- Kobayashi K, Asakura H, Shinozawa T, Yoshida S, Ichikawa Y, Tsuchiya M, Brown WR. Protein-losing enteropathy in systemic lupus erythematosus. Observations by magnifying endoscopy. Dig Dis Sci. 1989;34(12):1924-1928.
doi pubmed - Ebert EC. Gastrointestinal manifestations of Henoch-Schonlein Purpura. Dig Dis Sci. 2008;53(8):2011-2019.
doi pubmed - Choong CK, Beasley SW. Intra-abdominal manifestations of Henoch-Schonlein purpura. J Paediatr Child Health. 1998;34(5):405-409.
doi pubmed - Kato S, Ozawa K, Ando N, Naganuma H, Iinuma K, Nagura H. Immunoglobulin A enteropathy: a possible variant of Henoch-Schonlein purpura. Dig Dis Sci. 2004;49(11-12):1777-1781.
doi pubmed - Nakamura S, Hisamatsu T, Kikuchi J, Adachi M, Yamagishi Y, Imaeda H, Hosoe N, et al. A case of IgA-related enteropathy complicated with gastrointestinal bleeding and progressive IgA nephropathy: a possible variant Henoch-Schonlein purpura? Intern Med. 2010;49(16):1755-1761.
doi pubmed - Mills JA, Michel BA, Bloch DA, Calabrese LH, Hunder GG, Arend WP, Edworthy SM, et al. The American College of Rheumatology 1990 criteria for the classification of Henoch-Schonlein purpura. Arthritis Rheum. 1990;33(8):1114-1121.
doi pubmed - Ozen S, Pistorio A, Iusan SM, Bakkaloglu A, Herlin T, Brik R, Buoncompagni A, et al. EULAR/PRINTO/PRES criteria for Henoch-Schonlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Ann Rheum Dis. 2010;69(5):798-806.
doi pubmed - Tizard EJ, Hamilton-Ayres MJ. Henoch Schonlein purpura. Arch Dis Child Educ Pract Ed. 2008;93(1):1-8.
doi pubmed - Nagamori T, Oka H, Koyano S, Takahashi H, Oki J, Sato Y, Murono K, et al. Construction of a scoring system for predicting the risk of severe gastrointestinal involvement in Henoch-Schonlein Purpura. Springerplus. 2014;3:171.
doi pubmed - Fang Y, Peng K, Zhao H, Chen J. The characteristics of video capsule endoscopy in pediatric Henoch-Schonlein purpura with gastrointestinal symptoms. Pediatr Rheumatol Online J. 2020;18(1):84.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.