

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 14, Number 6, June 2023, pages 204-207

A Rare Case of Severe Jaundice in a Panhypopituitarism Patient
Jennifer Wiesea, Abdel Wahap El Ghezewia, Mujtaba Mohamedb, c, Tejas Joshib, Wesam Frandahb
aDepartment of Medicine, Marshall University Joan C. Edwards School of Medicine, Huntington, WV 25701, USA
bSection of Gastroenterology and Hepatology, Marshall University Joan C. Edwards School of Medicine, Huntington, WV 25701, USA
cCorresponding Author: Mujtaba Mohamed, Section of Gastroenterology and Hepatology, Marshall University Joan C. Edwards School of Medicine, Huntington, WV 25701, USA
Manuscript submitted April 20, 2023, accepted May 31, 2023, published online June 29, 2023
Short title: Rare Severe Jaundice in Panhypopituitarism
doi: https://doi.org/10.14740/jmc4102
| Abstract | ▴Top |
Hyperbilirubinemia and transaminitis are rarely associated with a disorder of endocrine function. It mostly manifests as a cholestatic pattern of liver injury. Herein, a 25-year-old female patient with a past medical history of congenital hypopituitarism due to pituitary ectopia presented with serum direct bilirubin level of 9.9 mg/dL and aspartate transaminase (AST)/alanine transaminase (ALT) of 60/47 U/L. All tests for chronic liver disease imaging and liver biopsy were normal. She was found to have central hypothyroidism and low cortisol level. She was started on intravenous (IV) levothyroxine 75 µg daily and IV hydrocortisone 10-5 mg AM/PM. She was discharged on oral levothyroxine 88 µg daily and hydrocortisone orally 10 mg twice daily. Follow-up labs 1 month later showed completely normal liver function test. In conclusion, hyperbilirubinemia due to congenital hypopituitarism can occur in adults. Delayed recognition of underlying endocrine disorder as a cause of hyperbilirubinemia and hepatocellular inflammation can result in end-stage liver damage due to prolonged cholestasis.
Keywords: Panhypopituitarism; Hyperbilirubinemia; Cholestasis
| Introduction | ▴Top |
Hyperbilirubinemia is defined as a serum total bilirubin greater than 1.2 mg/dL. Metabolism of bilirubin by the liver is comprised of uptake from the circulation, intracellular storage, conjugation with glucuronic acid, and biliary excretion. The pathophysiology of cholestasis in pituitary hormone insufficiency/deficiency is still unclear, but it is known that thyroid hormone, growth hormone (GH), and cortisol are involved independently in bile acid formation and secretion. Thyroid hormone and cortisol can influence bile acid-independent bile flow and formation. GH may also affect liver function as well as bile acid synthesis and secretion [1-3].
Herein, we report a case of a woman with congenital hypopituitarism who was non-compliant with her medications, presented with nausea, vomiting and hyperbilirubinemia and was found to have central hypothyroidism and adrenal insufficiency.
| Case Report | ▴Top |
A 25-year-old female patient with a past medical history of congenital hypopituitarism due to pituitary ectopia seen in magnetic resonance imaging (MRI) (Fig. 1) who lost follow-up with her endocrine doctor since age of 18 not on any hormone replacement therapy, history of depression and anxiety, amenorrhea, recurrent nausea and vomiting presented with a 4-day history of abdominal pain, nausea, vomiting, and dark urine. She denied any sick contacts, no new medications or herbals including over the counter medications. She was found to have jaundice on clinical exam. She had a dry skin texture on exam, thin hair, and hair patch loss including her eyebrows (Figs. 2, 3). Vital signs showed blood pressure of 100/50 mm Hg, heart rate of 110 bpm, height of 5.1 (155 cm), weight of 147 lbs, and body mass index (BMI) of 27 kg/m2. Laboratory data showed hemoglobin of 11.3 g/dL (reference range of 14 - 18 g/dL), platelet of 274 × 103/mm3 (150 - 440 × 103/mm3), mean corpuscular volume (MCV) of 86.7 fL (80 - 100 fL), red cell distribution width (RDW) of 43.8 fL (37 - 43 fL), albumin of 3.9 g/dL (3.1 - 4.5 g/dL), serum glucose of 74 mg/dL (74 - 100 mg/dL), serum potassium of 4 mEq/L (reference value: 3.1 - 3.5 mEq/L), serum sodium of 134 mEq/L (135 - 145 mEq/L), corticotropin (adrenocorticotropic hormone (ACTH)) level collected in the following morning of 3 pg/mL (7.2 - 63 pg/mL), luteinizing hormone (LH) of 1.3 mIU/mL (peak phase 22 - 76.1 mIU/mL), follicular-stimulating hormone (FSH) of 2.8 mIU/mL (midcycle peak level 5.2 - 17.5 mIU/mL), and serum prolactin of 19.9 ng/mL (2.2 - 30.3 ng/mL). She had a low level of thyroid-stimulating hormone (TSH) of 0.16 mIU/L (reference range of 0.35 - 3.7 mIU/L), free T4 level of 0.59 ng/dL (reference value 0.76 - 1.47 ng/dL), and serum cortisol of 0.5 µg/dL (reference value of 3.1 - 22.4 µg/dL). Aspartate transaminase (AST)/alanine transaminase (ALT) was 60/47 U/L (15 - 37/12 - 78 U/L) and serum total bilirubin was 9.9 mg/dL (reference 0.2 - 1.0 mg/dL) with direct bilirubin of 7.2 mg/dL. Total bilirubin continued to rise and peaked at 12 mg/dL. She had a normal iron profile, hepatitis panel, anti-smooth muscle antibody, anti-mitochondrial antibody, ceruloplasmin level, celiac panel, drug screen level, and human immunodeficiency virus (HIV) antibody. Abdominal ultrasound showed enlarged liver (17.4) with heterogenous liver texture, common bile duct diameter of 2 mm, no gallstones seen in gallbladder. Magnetic resonance cholangiopancreatography (MRCP) was negative for any biliary obstruction (Fig. 4). Endoscopic ultrasound did not reveal any abnormality. Patient had liver biopsy which was normal (Fig. 5). Patient was started on IV levothyroxine 75 µg daily and IV hydrocortisone 10-5 mg am/pm. Liver function started to improve with her serum bilirubin, AST, and ALT of 3.5 mg/dL, 33 U/L, and 27 U/L at the time of discharge. She was discharged on oral levothyroxine 88 µg daily and hydrocortisone orally 10 mg twice daily. Follow-up labs 1 month after discharge showed completely normal liver function test.
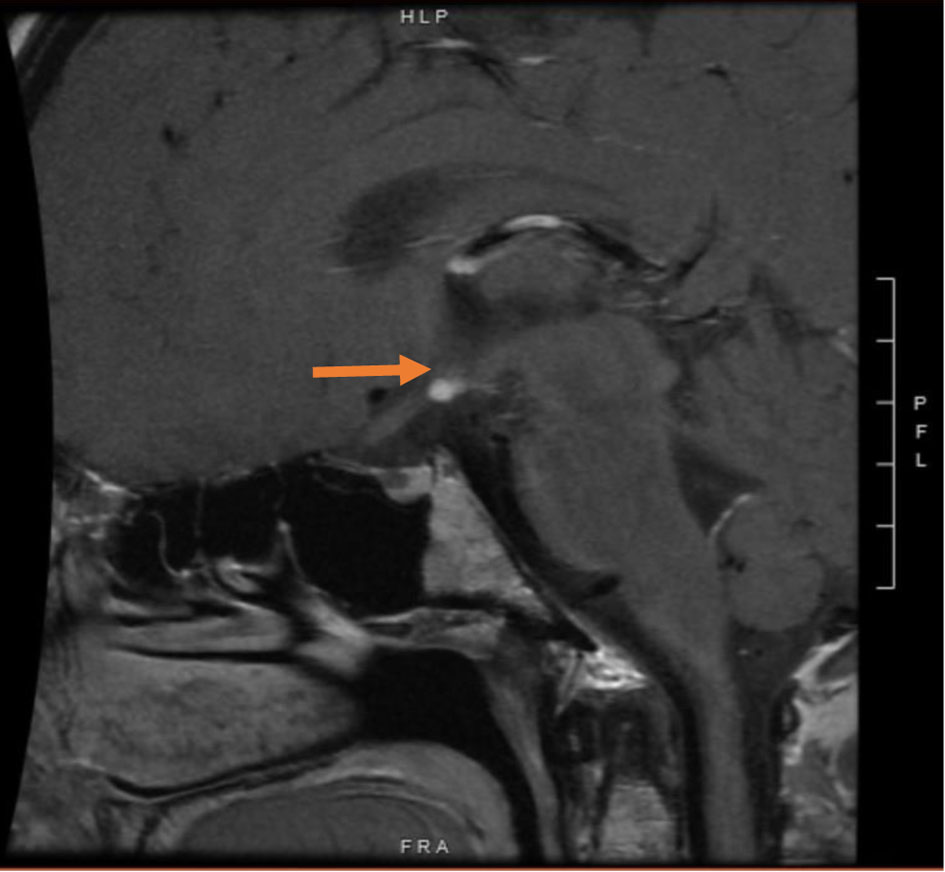 Click for large image | Figure 1. Magnetic resonance imaging of the brain showing ectopic posterior pituitary and stalk abnormality (arrow). |
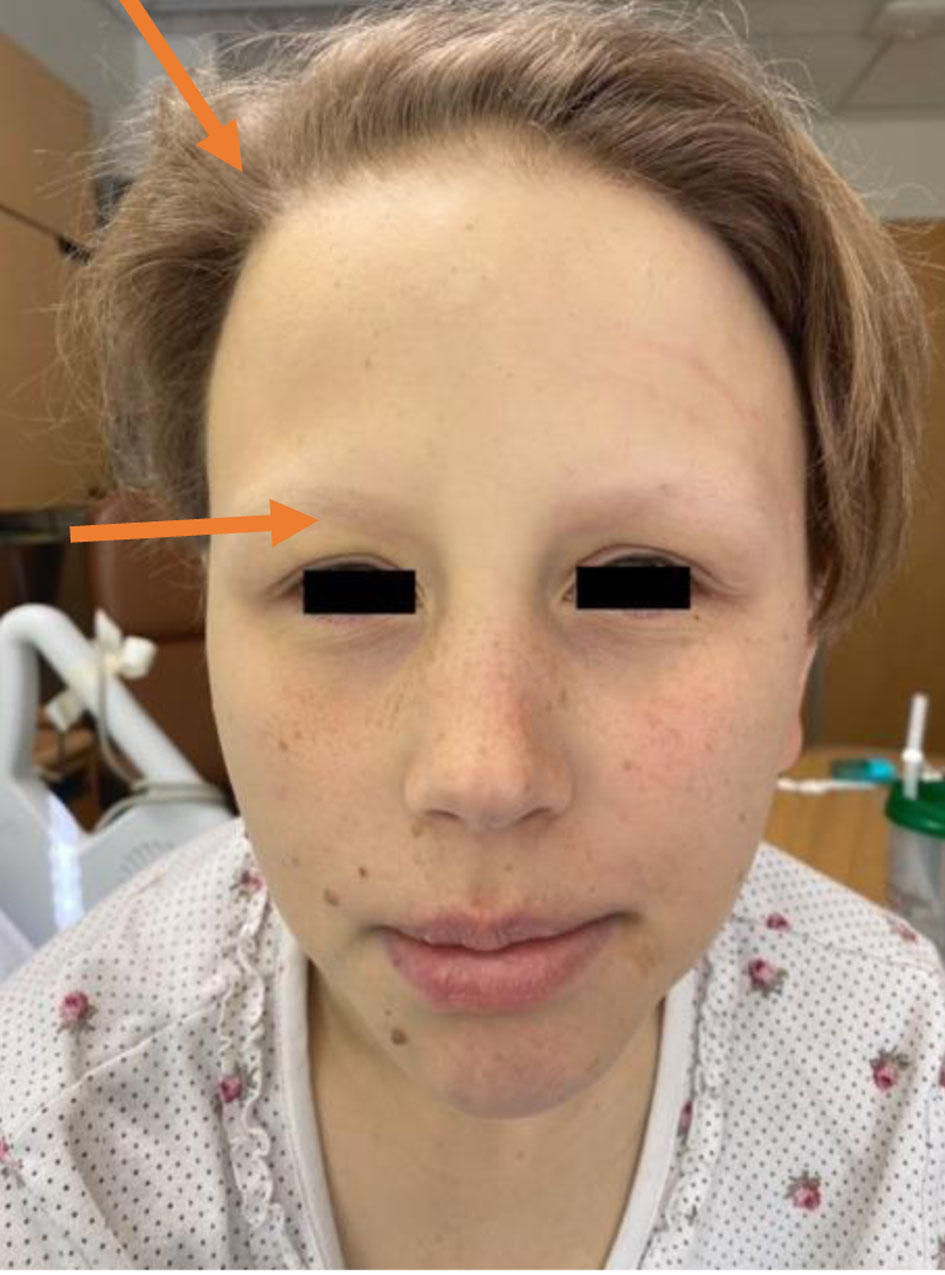 Click for large image | Figure 2. Thin hair in the scalp and loss of hair in the eyebrows (arrows). |
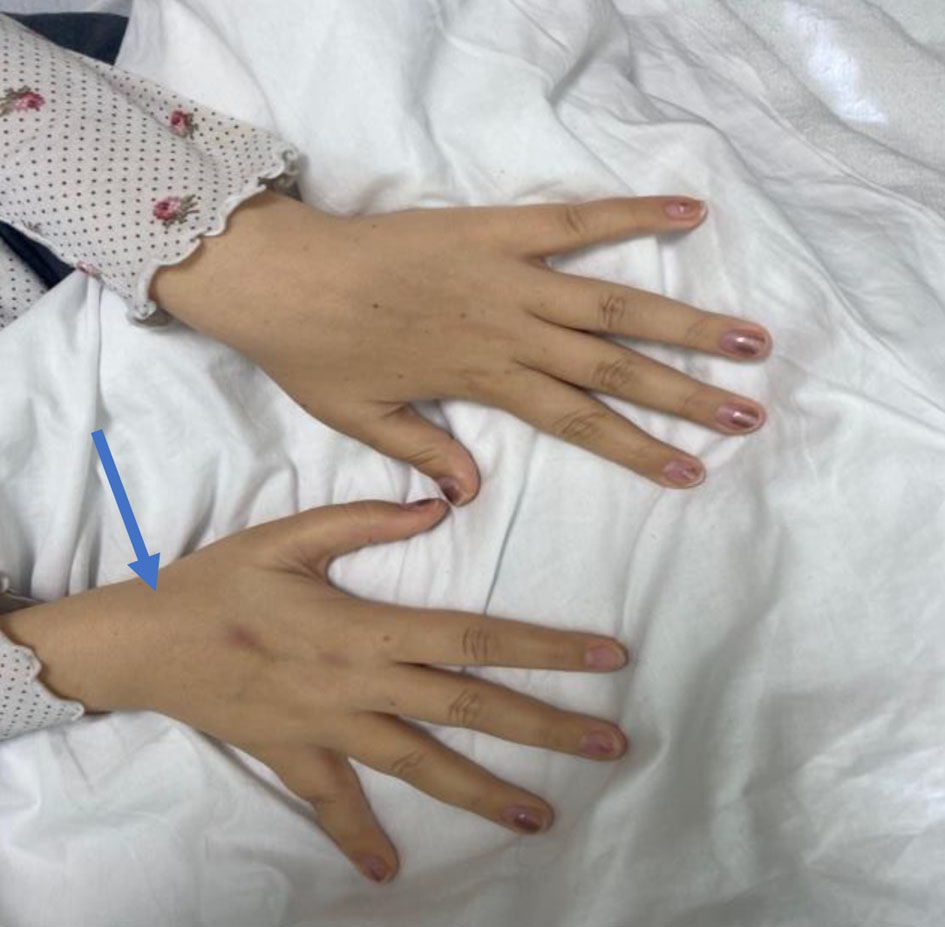 Click for large image | Figure 3. The dry skin texture on the dorsum of the hand. |
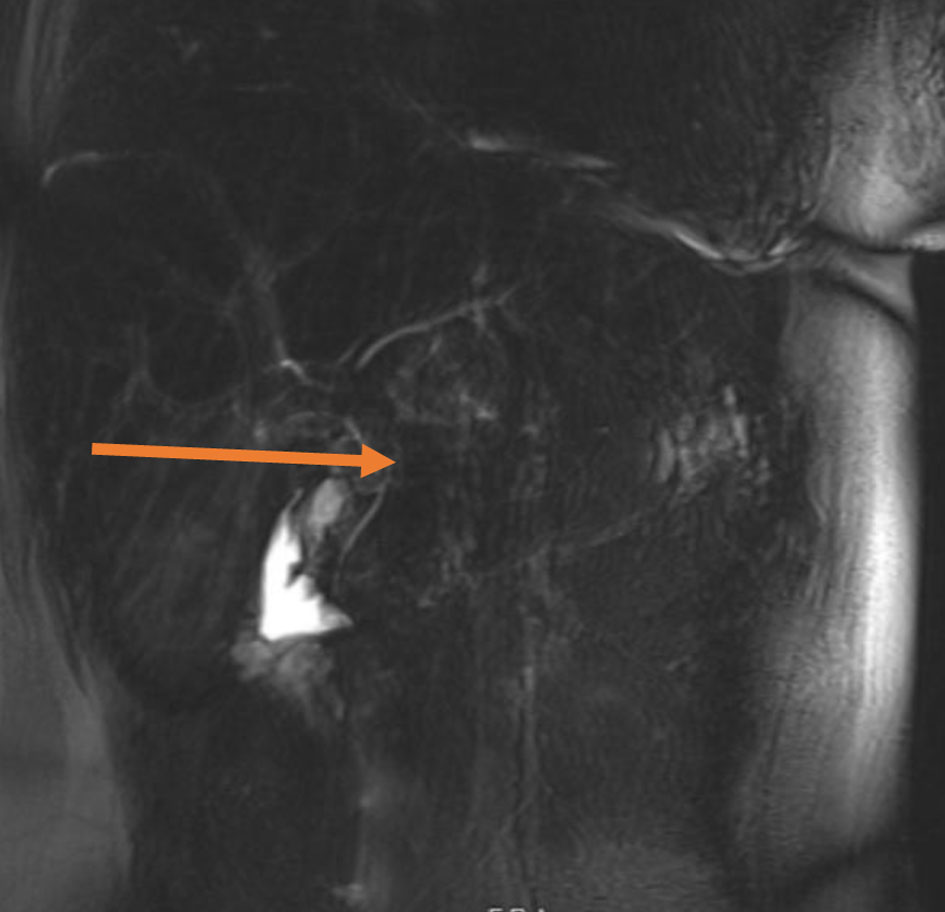 Click for large image | Figure 4. Magnetic resonance cholangiopancreatography showing extrahepatic bile duct without dilation (arrow). |
 Click for large image | Figure 5. Normal liver biopsy (arrow). |
| Discussion | ▴Top |
Congenital hypopituitarism is defined as the deficiency of one or more hormones produced by the anterior or posterior pituitary gland. Its incidence is estimated to be between 1 in 4,000 and 1 in 10,000 live births [4]. Ectopic posterior pituitary (EPP) can occur with isolated growth hormone deficiency (IGHD) or combined pituitary hormone deficiency (CPHD) [5]. It is believed that it is caused by certain genetic mutations. However, no exact gene was identified. Isolated reports of mutations in the HESX1, LHX4 and SOX3 [6-8] have been associated with the development of an EPP. It is reported that endocrine disorders such as hypopituitarism or hypothyroidism are rare (2%) in the causes of infantile cholestasis [9]. A very small number of cases in adults have been described in the literature [10]. It is very well known that there is a strong association between liver injury and hypophyseal hormonal deficiencies [11]. Pathogenesis process is not fully understood. Karnsakul et al reported that hepatitis could be secondary to a deficit in cortisol and/or GH, which participates in the regulation, synthesis, and transport of biliary acids [12]. Others reported that TSH deficiency affects canalicular bile secretion, probably by alterations in the Na+/K+ ATPase activity in the plasma membrane of hepatocytes [12]. Moreover, adenohypophyseal hormone deficiency produces abnormalities of bile canalicular structure, essential for conjugated bilirubin excretion [12]. But in general deficiency of one or more pituitary hormones delays the maturation of the transport mechanisms of conjugated bilirubin causing dysfunction of the biliary channels and resulting in its accumulation which eventually result in cholestasis and jaundice. In a long time, this can result in end-stage liver disease [10]. In our patient, autoimmune and obstructive pathology was ruled out based on serology, imaging studies and tissue biopsy.
Liver damage may persist if diagnosis and hormone replacement therapy is delayed [11]. End-stage liver disease did not occur in this case as the patient used to receive hormone replacement therapy and stopped taking them for few years before this presentation. In this patient, there was no evidence that end-stage liver damage only increased echogenicity in liver ultrasound without evidence of portal hypertension in terms of ascites, varices or portal hypertension gastropathy on endoscopy. According to case series by Spray et al, most of these liver chemistry abnormalities are reversible with hormonal replacement therapy [11]. In that case series, 12 children had neonatal hepatitis and hypopituitarism. Hepatitis and abnormal liver enzymes were resolved in nine of 12 children within 6 weeks after treatment with thyroxine, hydrocortisone, and GH [8]. In our patient, her liver enzymes normalized 5 weeks after starting hormone replacement therapy.
Conclusion
Although a couple of cases of hyperbilirubinemia were documented in association with congenital hypopituitarism in neonates and infants, only a few cases in adults have been reported. Delayed detection of underlying endocrine disorder as a cause of hyperbilirubinemia and hepatocellular inflammation can result in substantial morbidity and mortality. Early diagnosis and treatment of endocrine disorder can consequently reduce morbidity and mortality. This case can remind clinicians of the importance of evaluating pituitary and endocrine disorder in patients with presentation of hyperbilirubinemia and transaminitis after common causes are ruled out.
Learning points
Thorough history, examination and interpretation of laboratory data are essential to early diagnose cases of direct hyperbilirubinemia secondary to panhypopituitarism. Diagnosis can be challenging as initial labs can be misleading in central hypopituitarism.
Acknowledgments
We would like to acknowledge our library staff at Cabell Huntington Hospital for supplying us with articles to complete this manuscript.
Financial Disclosure
This project was not supported by any grant or funding agency.
Conflict of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Informed Consent
The patient described in the case report had given informed consent for the case report to be published.
Author Contributions
Each author has individually been involved and participated in drafting the manuscript and revising it critically for important intellectual content and have given final approval of the version to be published. Each has agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. WF and TJ encouraged JW, AE, and MM to learn about panhypopituitarism, mechanism of liver injury in those patients. All authors discussed the medical literature. JW and AE presented the idea, and JW, MM, AE wrote the manuscript with input from all authors.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Fawaz R, Baumann U, Ekong U, Fischler B, Hadzic N, Mack CL, McLin VA, et al. Guideline for the evaluation of cholestatic jaundice in infants: joint recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr. 2017;64(1):154-168.
doi pubmed - Ellaway CJ, Silinik M, Cowell CT, Gaskin KJ, Kamath KR, Dorney S, Donaghue KC. Cholestatic jaundice and congenital hypopituitarism. J Paediatr Child Health. 1995;31(1):51-53.
doi pubmed - DeSalvo D, Pohl JF, Wilson DP, Bryant W, Easley D, Greene J, Santiago J. Cholestasis secondary to panhypopituitarism in an infant. J Natl Med Assoc. 2008;100(3):342-344.
doi pubmed - Bando H, Urai S, Kanie K, Sasaki Y, Yamamoto M, Fukuoka H, Iguchi G, et al. Novel genes and variants associated with congenital pituitary hormone deficiency in the era of next-generation sequencing. Front Endocrinol (Lausanne). 2022;13:1008306.
doi pubmed pmc - Hamilton J, Blaser S, Daneman D. MR imaging in idiopathic growth hormone deficiency. AJNR Am J Neuroradiol. 1998;19(9):1609-1615.
pubmed pmc - Sloop KW, Walvoord EC, Showalter AD, Pescovitz OH, Rhodes SJ. Molecular analysis of LHX3 and PROP-1 in pituitary hormone deficiency patients with posterior pituitary ectopia. J Clin Endocrinol Metab. 2000;85(8):2701-2708.
doi pubmed - Carvalho LR, Woods KS, Mendonca BB, Marcal N, Zamparini AL, Stifani S, Brickman JM, et al. A homozygous mutation in HESX1 is associated with evolving hypopituitarism due to impaired repressor-corepressor interaction. J Clin Invest. 2003;112(8):1192-1201.
doi pubmed pmc - Machinis K, Pantel J, Netchine I, Leger J, Camand OJ, Sobrier ML, Dastot-Le Moal F, et al. Syndromic short stature in patients with a germline mutation in the LIM homeobox LHX4. Am J Hum Genet. 2001;69(5):961-968.
doi pubmed pmc - Gottesman LE, Del Vecchio MT, Aronoff SC. Etiologies of conjugated hyperbilirubinemia in infancy: a systematic review of 1692 subjects. BMC Pediatr. 2015;15:192.
doi pubmed pmc - Valle-Murillo MA, Perez-Diaz I. Hepatopathy in an adult, secondary to congenital untreated panhypopituitarism and ectopic posterior pituitary gland. Indian J Endocrinol Metab. 2012;16(5):821-823.
doi pubmed pmc - Spray CH, McKiernan P, Waldron KE, Shaw N, Kirk J, Kelly DA. Investigation and outcome of neonatal hepatitis in infants with hypopituitarism. Acta Paediatr. 2000;89(8):951-954.
doi pubmed - Karnsakul W, Sawathiparnich P, Nimkarn S, Likitmaskul S, Santiprabhob J, Aanpreung P. Anterior pituitary hormone effects on hepatic functions in infants with congenital hypopituitarism. Ann Hepatol. 2007;6(2):97-103.
pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.