

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 14, Number 7, July 2023, pages 265-269

There’s More Than Meets the Eye: Wolfram Syndrome in a Type I Diabetic Patient
Jasvindar Kumara, Atif Ahmedb, Mashal Khanc, d, Yasir Ahmedd, e
aInternal Medicine at Basset Medical Center, Cooperstown, NY, USA
bDepartment of Psychiatry, Khyber Medical University, Peshawar, Pakistan
cKhyber Medical University, Peshawar, Pakistan
dDepartment of Internal Medicine, United Health Services Hospitals, Binghamton, NY, USA
eCorresponding Author: Yasir Ahmed, Department of Internal Medicine, United Health Services Hospitals, Binghamton, NY 13903, USA
Manuscript submitted June 10, 2023, accepted July 27, 2023, published online July 31, 2023
Short title: Wolfram Syndrome in a Type I Diabetic Patient
doi: https://doi.org/10.14740/jmc4128
| Abstract | ▴Top |
Wolfram syndrome (WS) is a rare neurodegenerative and genetic disorder, also known by the synonym DIDMOAD, which stands for diabetes insipidus (DI), childhood-onset diabetes mellitus (DM), optic atrophy (OA), and deafness (D). We present a case of a 25-year-old diabetic patient, using insulin for 15 years, who had increasing polyuria and polydipsia, along with progressive hearing and vision loss. Laboratory tests revealed elevated hemoglobin A1c (HbA1c) and blood sugar levels. Optic nerve, optic chiasm, pons, and brain stem atrophy was seen on magnetic resonance imaging (MRI) of brain. After workup, a diagnosis of DI was made. Once the diagnosis was reached, treatment with subcutaneous insulin and nasal desmopressin improved patient’s symptoms. In juvenile diabetic patients presenting with new onset or worsening polyuria and polydipsia, the possibility of WS should be considered. Early diagnosis and initiation of appropriate management leads to improved outcomes and the quality of life.
Keywords: Wolfram syndrome; Diabetes type I; Optic atrophy; Deafness; DIDMOAD
| Introduction | ▴Top |
DIDMOAD is the acronym often used synonymously for Wolfram syndrome (WS), which includes the four characteristic clinical abnormalities of diabetes insipidus (DI), diabetes mellitus (DM), optic atrophy (OA), and deafness (D) [1, 2]. Out of the four characteristics mentioned, childhood-onset DM and OA are considered sufficient for diagnosing WS. It is now considered a multi-system neurodegenerative disorder that can have symptoms of ataxia, behavioral and psychiatric illness, apnea, hypogonadism, and renal tract atonia with hydronephrosis [2].
Diagnosis is often delayed because polyuria and polydipsia are usually attributed to poorly controlled diabetes, while visual symptoms are thought to be a consequence of diabetic retinopathy. A delay of at least 7 years occurred in a cohort of pediatric patients with diabetes before WS was recognized [3]. The management of WS is different from classic type I diabetes, hence, recognition of WS in diabetic patients is very important for improving outcomes. We are sharing our experience of WS in a young diabetic patient whose polyuria, polydipsia, and vision problems were attributed to poorly controlled type I diabetes.
| Case Report | ▴Top |
Investigations
A 25-year-old diabetic female patient presumed to have type I DM, managed with insulin for almost 15 years, presented with increasing polyuria and polydipsia for 1 month. No prior records were available to confirm the diagnosis of type I DM. She reported that despite drinking more than 10 L of water in the 24 h prior to presentation, her thirst was not relieved. Polyuria was not associated with dysuria, hematuria or pyuria. She denied having headache, galactorrhea, sexual dysfunction, weight changes, amenorrhea, and heat or cold intolerance. Upon further questioning of review of symptoms, she mentioned gradually progressive hearing and vision loss. Vital signs were stable, and tongue and oral mucosa were slightly dry on physical examination. Cranial nerve examination revealed loss of peripheral vision and sensorineural hearing. The rest of the examination was unremarkable.
Diagnosis
Her symptoms were initially attributed to poorly controlled diabetes, and consideration was given to primary polydipsia, and central or nephrogenic DI. Initial laboratory workup showed random blood glucose level of 221 mg/dL, and hemoglobin A1c (HbA1c) of 10%, while electrolytes, kidney function and liver profile were within normal limits. Blood urea nitrogen (BUN) and creatinine (Cr) were also within the normal range. Initially, the serum osmolality was 296 mmol/kg of H2O (275 - 295 mOsm/kg H2O), and spot urine osmolality was 117 mmol/kg (50 to 1,200 mOsm/kg of H2O). The urine concentration remained the same after water deprivation test, ruling out primary polydipsia. The serum and urine osmolalities improved to 270 and 650 mmol/kg of H2O, respectively after desmopressin administration. Ultrasound of abdomen and pelvis was normal. Magnetic resonance imaging (MRI) of the brain revealed optic nerve, optic chiasma, pons, and brain stem atrophy. Aside from loss of intrinsic T1 high signal of the neurohypophysis, the pituitary was of normal size (Figs. 1 and 2). The presence of DI in a patient with childhood-onset DM and progressive vision and hearing loss led to the diagnosis of WS.
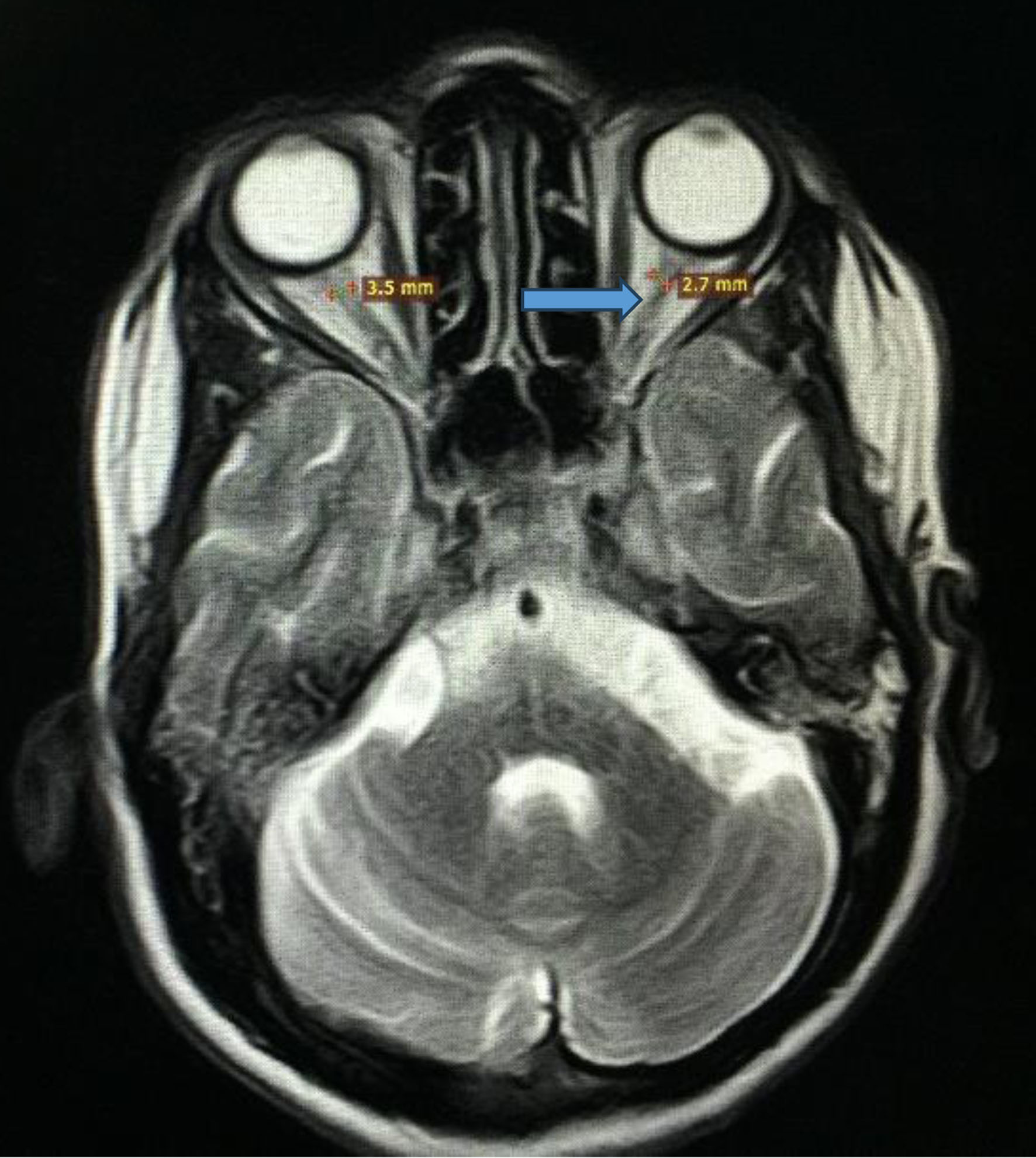 Click for large image | Figure 1. Magnetic resonance imaging of brain and orbits showing optic nerve atrophy as indicated by the blue arrow. |
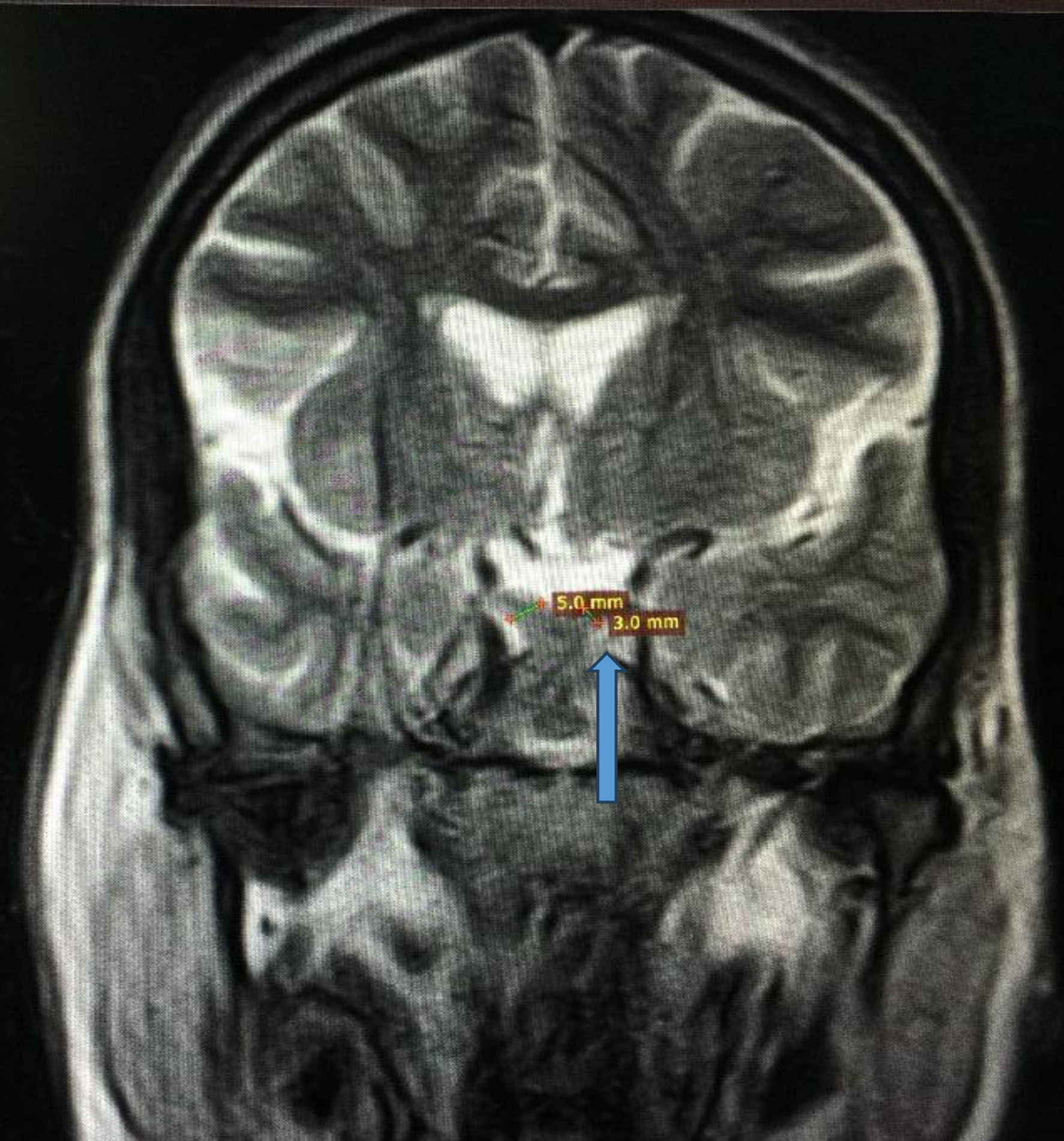 Click for large image | Figure 2. Magnetic resonance imaging of brain showing optic chiasm atrophy (3 mm vs. 5 mm) indicated by the blue arrow. |
Treatment
Treatment was initiated with subcutaneous insulin and nasal desmopressin. In-house ophthalmology and maxillofacial (ear, nose, throat) consultations were placed for vision and hearing evaluation, as well as outpatient follow-up.
Follow-up and outcomes
Polyuria and polydipsia resolved in response to treatment. She was extensively counseled and educated on diet, regular physical activity, insulin administration, glucose monitoring, and medication adherence. She is responding positively to insulin and desmopressin.
| Discussion | ▴Top |
WS is a rare neurodegenerative and genetic disorder, estimated to afflict about 1 in 160,000 - 770,000 [1, 2, 4]. Wolfram and Wagener first reported it when they found four of eight siblings with juvenile DM and OA [5]. A study of 68 patients reported that the onset of type I DM is the first manifestation of WS (median age of diagnosis: 6.5 years), followed by OA (at 13 years), marked by loss of color and peripheral vision [4]. Another study reported the median ages of onset of type I DM and OA as 6 and 11 years, respectively [2]. Central DI (affecting 70% of patients) and sensorineural deafness (affecting 65% of patients) are reported to occur in the second decade. Hearing problems can range in severity from deafness beginning at birth, to mild hearing loss beginning in adolescence that worsens over time [2, 4]. Renal-tract abnormalities affecting 70% of patients have been reported as early as 7 years of age [1, 4], but more commonly occur in the second or third decade [1, 2, 4]. Along with neurologic, pituitary-gonadal, cardiac, and gastrointestinal complications, even congenital malformations have been reported in WS to varying degrees, occurring most often late in the disease [2].
Recessive mutations in the Wolfram syndrome 1 gene (WFS1) usually confirm the diagnosis in the majority of patients. However, several dominant mutations have also been discovered [1, 6]. A small number of patients carry recessive mutations in the CISD2 (WFS2) gene [7]. WS occurs due to the endoplasmic reticulum (ER) dysfunction. Mutations in the WFS1 gene which encodes a transmembrane protein localized to the ER cells and neuronal cells lead to elevated ER stress levels, and the initiation of ER stress-associated cell death [8, 9]. Although there are no definitive treatments to slow down or reverse the disease progression in WS, supportive care and regular monitoring can improve the quality of life. Patients are maintained on insulin for better glycemic control, while DI is treated with desmopressin. Polysomnography, annual neurologic examination, eye examination, and audiometry are recommended. Prescription glasses, hearing aids, and cochlear implant address the vision and hearing problems to some extent. Ataxia, dysarthria, and dysphagia are common neurologic complaints. A speech and language pathologist is helpful in that regard. Brain stem atrophy which usually leads to death by central apnea is another prominent feature and management requires taking a pulmonologist on board [10].
Novel investigational drugs that target ER, such as 4-phenyl butyric acid and tauroursodeoxycholic acid (chemical chaperones that stabilize protein conformation during folding and improve trafficking), and dantrolene (targets ryanodine receptors localized to the ER membrane), can prevent the death of neurons and β cells in WS [11, 12]. Other novel treatments include gene therapy by using clustered regularly interspaced short palindromic repeats (CRISPR) and mesencephalic astrocyte-derived neurotrophic factor (MANF), which can prevent cell death and activate the proliferation of affected cells in WS [13, 14]. Regenerative medicine can be used to create induced pluripotent stem (iPS) cells using patients’ skin cells, correcting the WFS1 gene mutations with genome editing technology, and differentiate these iPS cells into insulin-producing β cells, retinal cells, and neurons for transplantation [15]. It has been shown that glucagon-like peptide-1 receptor (GLP-1R) agonists can improve diabetes and suppress apoptosis in cell models of WS [16]. It is believed that understanding the pathogenesis will not only help in the development of novel therapeutics for WS, but also help treat similar diseases caused by ER dysfunction, as its main underlying pathology lies in dysfunction of the ER.
WS carries a poor prognosis, with the median age of death being 30 years (range 25 - 49 years), usually resulting from respiratory failure as a consequence of brain stem atrophy [2]. The most common causes of morbidity and mortality are the neurological manifestations of this syndrome and the complications of urinary tract atony [4].
Conclusion
Symptoms like polyuria, polydipsia, and vision loss can easily be attributed to poorly controlled diabetes. Early identification of WS is very important as the management of WS differs from DM and mortality for WS is higher than classic DM. This case highlights early recognition of WS and the provision of timely appropriate interventions to improve the quality of life, by involving a multi-disciplinary care team that includes social workers and welfare organizations as well as a long-term follow-up plan. The need for further studies and including trials to improve treatment options is also recommended.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Informed consent has been obtained.
Author Contributions
Jasvindar Kumar, primary author, contributed to data collection, literature review and drafting the manuscript. Atif Ahmed contributed to introduction and discussion, drafting the manuscript. Mashal Khan contributed to discussion, drafting, review and editing and referencing. Yasir Ahmed supervised the entire process, from conception to finalizing the draft, and contributed to literature search, review and editing and final draft for publication.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Hansen L, Eiberg H, Barrett T, Bek T, Kjaersgaard P, Tranebjaerg L, Rosenberg T. Mutation analysis of the WFS1 gene in seven Danish Wolfram syndrome families; four new mutations identified. Eur J Hum Genet. 2005;13(12):1275-1284.
doi pubmed - Barrett TG, Bundey SE, Macleod AF. Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet. 1995;346(8988):1458-1463.
doi pubmed - Zmyslowska A, Borowiec M, Fichna P, Iwaniszewska B, Majkowska L, Pietrzak I, Szalecki M, et al. Delayed recognition of Wolfram syndrome frequently misdiagnosed as type 1 diabetes with early chronic complications. Exp Clin Endocrinol Diabetes. 2014;122(1):35-38.
doi pubmed - Kinsley BT, Swift M, Dumont RH, Swift RG. Morbidity and mortality in the Wolfram syndrome. Diabetes Care. 1995;18(12):1566-1570.
doi pubmed - Wolfram DJ, Wagener HP. Diabetes mellitus and simple optic atrophy among siblings: report of four cases. Mayo Clin Proc. 1938;1:715-718
- Inoue H, Tanizawa Y, Wasson J, Behn P, Kalidas K, Bernal-Mizrachi E, Mueckler M, et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet. 1998;20(2):143-148.
doi pubmed - Amr S, Heisey C, Zhang M, Xia XJ, Shows KH, Ajlouni K, Pandya A, et al. A homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. Am J Hum Genet. 2007;81(4):673-683.
doi pubmed pmc - Fonseca SG, Fukuma M, Lipson KL, Nguyen LX, Allen JR, Oka Y, Urano F. WFS1 is a novel component of the unfolded protein response and maintains homeostasis of the endoplasmic reticulum in pancreatic beta-cells. J Biol Chem. 2005;280(47):39609-39615.
doi pubmed - Fonseca SG, Ishigaki S, Oslowski CM, Lu S, Lipson KL, Ghosh R, Hayashi E, et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J Clin Invest. 2010;120(3):744-755.
doi pubmed pmc - Urano F. Wolfram syndrome: diagnosis, management, and treatment. Curr Diab Rep. 2016;16(1):6.
doi pubmed pmc - Shang L, Hua H, Foo K, Martinez H, Watanabe K, Zimmer M, Kahler DJ, et al. beta-cell dysfunction due to increased ER stress in a stem cell model of Wolfram syndrome. Diabetes. 2014;63(3):923-933.
doi pubmed pmc - Lu S, Kanekura K, Hara T, Mahadevan J, Spears LD, Oslowski CM, Martinez R, et al. A calcium-dependent protease as a potential therapeutic target for Wolfram syndrome. Proc Natl Acad Sci U S A. 2014;111(49):E5292-5301.
doi pubmed pmc - Petrova P, Raibekas A, Pevsner J, Vigo N, Anafi M, Moore MK, Peaire AE, et al. MANF: a new mesencephalic, astrocyte-derived neurotrophic factor with selectivity for dopaminergic neurons. J Mol Neurosci. 2003;20(2):173-188.
doi pubmed - Lindahl M, Danilova T, Palm E, Lindholm P, Voikar V, Hakonen E, Ustinov J, et al. MANF is indispensable for the proliferation and survival of pancreatic beta cells. Cell Rep. 2014;7(2):366-375.
doi pubmed pmc - Urano F. Wolfram syndrome iPS cells: the first human cell model of endoplasmic reticulum disease. Diabetes. 2014;63(3):844-846.
doi pubmed pmc - Kondo M, Tanabe K, Amo-Shiinoki K, Hatanaka M, Morii T, Takahashi H, Seino S, et al. Activation of GLP-1 receptor signalling alleviates cellular stresses and improves beta cell function in a mouse model of Wolfram syndrome. Diabetologia. 2018;61(10):2189-2201.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.