

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 14, Number 11, November 2023, pages 369-377

Stepwise Treatment for TAFRO Syndrome
Makoto Idea, e ![]() , Tomoko Yokoyamab, Masashi Ishikawac, Kazuki Kojimad
, Tomoko Yokoyamab, Masashi Ishikawac, Kazuki Kojimad
aDepartment of Hematology, Takamatsu Red Cross Hospital, Takamatsu, Kagawa, Japan
bDepartment of Nephrology, Takamatsu Red Cross Hospital, Takamatsu, Kagawa, Japan
cDepartment of Pathology, Takamatsu Red Cross Hospital, Takamatsu, Kagawa, Japan
dTakamatsu Red Cross Hospital Post Graduate Clinical Training Center, Takamatsu Red Cross Hospital, Takamatsu, Kagawa, Japan
eCorresponding Author: Makoto Ide, Department of Hematology, Takamatsu Red Cross Hospital, Takamatsu, Kagawa 760-0017, Japan
Manuscript submitted September 18, 2023, accepted November 2, 2023, published online November 23, 2023
Short title: Stepwise Treatment for TAFRO Syndrome
doi: https://doi.org/10.14740/jmc4160
| Abstract | ▴Top |
TAFRO syndrome, a rapidly progressive and fatal disease, is rare, and its etiology remains unknown. It is characterized by thrombocytopenia, anasarca (edema, pleural effusion, and ascites), fever, reticulin fibrosis (or renal insufficiency), and organomegaly with Castleman disease (CD)-like histological features in the lymph nodes. CD is a rare, indolent, lymphoproliferative disorder with no established curative strategies. Most idiopathic multicentric CD cases are controlled with anti-interleukin (IL)-6 therapy (tocilizumab and siltuximab) and/or rituximab. However, it is unclear whether these therapies can be directly applied to treat TAFRO syndrome. Here, we describe stepwise immunotherapy (rituximab induction therapy and cyclosporine maintenance therapy) for two cases of steroid-refractory TAFRO syndrome. A 32-year-old man visited a local hospital with sudden onset of fever and epigastralgia. The diagnosis of TAFRO syndrome was established based on the diagnostic criteria. After rituximab administration, C-reactive protein and IL-6 levels were normalized. However, the ascites persisted, with increased resistance to rituximab. Tocilizumab was also ineffective; therefore, cyclosporine was administered. After the initiation of cyclosporine treatment, the ascites decreased and ultimately disappeared. Twelve months after immunotherapy, the patient remained asymptomatic under cyclosporine maintenance therapy. Similar stepwise immunosuppressive therapy was administered to a 72-year-old man with TAFRO syndrome complicated by renal failure. After rituximab infusion, C-reactive protein was decreased. Although methylprednisolone, rituximab, tocilizumab, and cyclosporine were administered, other laboratory data and clinical symptoms remained unchanged. His level of consciousness subsequently deteriorated due to herpes zoster encephalitis, and he died. We consider the combination of rituximab induction therapy and cyclosporine maintenance therapy to be effective for TAFRO syndrome if initiated at an early stage.
Keywords: TAFRO syndrome; Castleman disease; Immunotherapy
| Introduction | ▴Top |
Castleman disease (CD) is a rare indolent lymphoproliferative disorder for which there is no established curative strategy [1]. The clinical variations include unicentric CD (UCD) (involving only a single lymph node station) and multicentric CD (MCD) (involving multiple lymph node stations and/or organs). The first case of UCD was reported by Castleman in 1956 [2]. MCD was initially described in 1978 [3] and was subsequently confirmed in a case series study [4]. There are currently two recognized categories of MCD: human herpes virus 8 (HHV-8)-associated MCD and idiopathic MCD (iMCD). HHV-8-associated MCD is caused by HHV-8 infection and occurs almost exclusively in the human immunodeficiency virus (HIV)-positive population [1]. CD was diagnosed using diagnostic criteria based on the pathological features of the lymph nodes, clinical symptoms, and laboratory data [5].
The severity of MCD varies from severe to mild and may progress from abnormal laboratory results to multiorgan failure. The majority of CD cases show a slowly progressive disease course that does not improve.
TAFRO syndrome is a rapidly progressive fatal disease that is very rare, has an unknown etiology, and is characterized by thrombocytopenia, anasarca (edema, pleural effusion, and ascites), fever, reticulin fibrosis (or renal insufficiency), and organomegaly [6, 7]. TAFRO syndrome was diagnosed using diagnostic criteria based on symptoms and laboratory data [7].
The lymph node swelling was generally mild and could not be biopsied. If biopsy is possible, the lymph node histology of TAFRO syndrome shows CD-like features and has been divided into hypervascular types, a new histological type of CD [6].
The TAFRO syndrome is generally considered a severe iMCD subtype. iMCD has also been classified as iMCD-not otherwise specified (NOS) and TAFRO-iMCD; however, these classifications have not been confirmed. Furthermore, it has not yet been established whether TAFRO syndrome is a severe subtype of CD or a CD-like independent disease [8, 9]. The overall survival was significantly longer in patients with iMCD-NOS than in those with TAFRO syndrome [9].
Recently, the treatment guidelines for UCD and iMCD were established to improve patient care [10, 11]. In accordance with these guidelines, anti-interleukin (IL)-6 therapy (tocilizumab and siltuximab), and rituximab have been used worldwide as first-line therapies in the MCD [1]. However, it is unclear whether these guidelines can be directly applied to the TAFRO syndrome.
Although a curative strategy for TAFRO syndrome has not yet been established, treatment options have recently been described. High-dose glucocorticoid therapy is the first-line treatment for TAFRO syndrome. If this treatment is ineffective, rituximab, tocilizumab, and cyclosporine are used as second-line treatment [9]. However, the sequence of these treatments and timing of their application remains unclear.
In an analysis of the first-line treatment for TAFRO syndrome, the time to the next treatment or death was significantly longer in rituximab-treated patients than in tocilizumab- or cyclosporine-treated patients [12]. This indicates that anti-IL-6 therapy is not as effective for TAFRO syndrome as it is for IMCD. Therefore, several therapies have been developed for the TAFRO syndrome.
In summary, our study aims to explore the effectiveness of a new stepwise immunotherapy approach for refractory TAFRO syndrome, shedding light on potential treatment options and contributing to the ongoing efforts to improve the management of this challenging condition.
| Case Reports | ▴Top |
Case 1
Investigations
A 32-year-old male patient who had been in good health visited a local hospital with sudden onset of epigastralgia. The patient had a family history of rheumatoid arthritis (mother and older brother) and adult Still’s disease (father). Neither pleural effusion nor ascites were detected on chest and abdominal computed tomography (CT), and gastrointestinal fiberscopy showed no abnormal findings. However, epigastralgia and pyrexia persisted for 1 month. One month after the first visit, the patient underwent re-examination to identify the cause of fever.
Diagnosis
Whole-body CT revealed bilateral ascites, pleural effusion, anasarca, generalized lymphadenopathy, and hepatosplenomegaly (Fig. 1a). Axillary lymph node biopsy revealed atrophied germinal centers with enlarged endothelial cell nuclei and proliferation of endothelial venules. These histological features are consistent with those of TAFRO-iMCD. Bone marrow biopsy showed the absence of myelofibrosis. Based on these criteria, the patient was diagnosed with TAFRO-iMCD [7] and was referred to our hospital for further treatment. The diagnostic criteria are summarized in Table 1 and are compared with those in case 2.
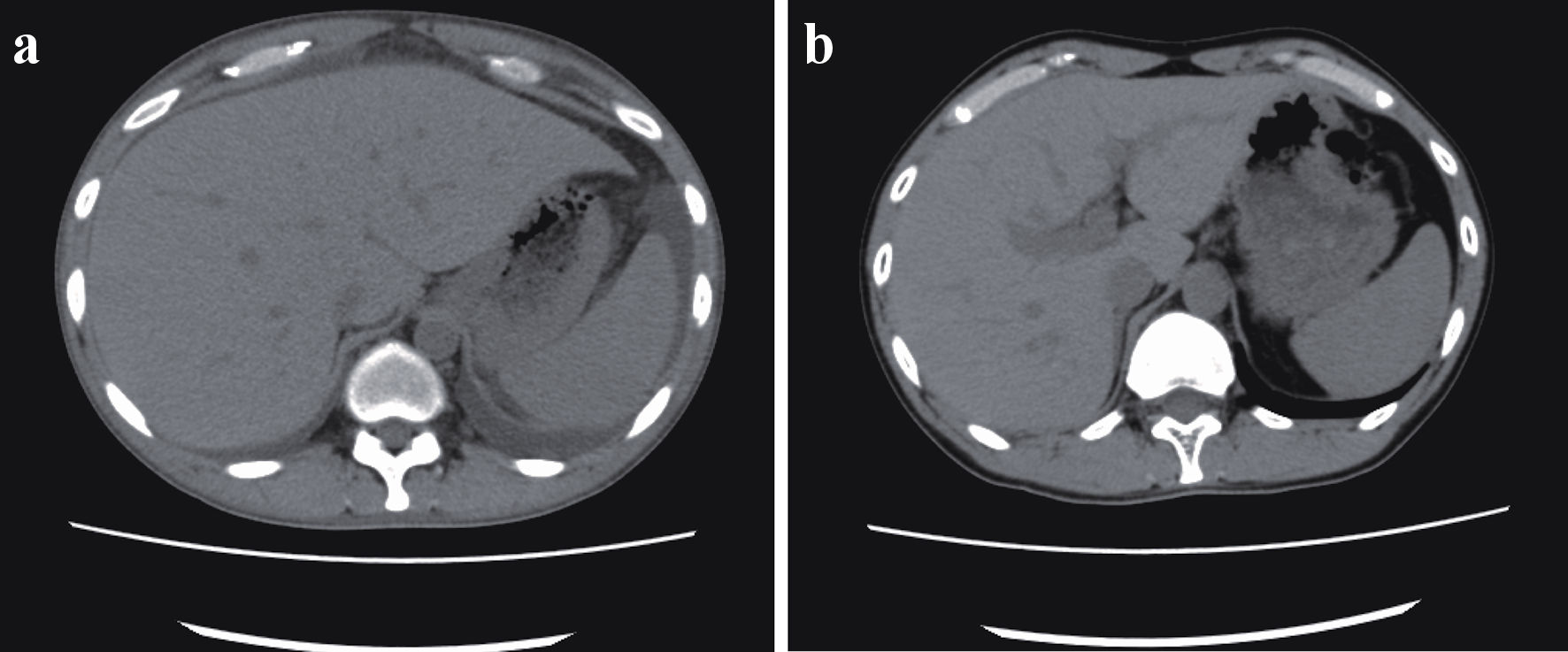 Click for large image | Figure 1. (a) Ascites before cyclosporine therapy. Whole-body computed tomography revealed bilateral ascites, pleural effusion, anasarca, generalized lymphadenopathy, and hepatosplenomegaly before the initiation of cyclosporine therapy. (b) Twelve months after cyclosporine therapy. After 12 months of cyclosporine maintenance therapy, the patient remained asymptomatic, without ascites or hepatosplenomegaly. |
 Click to view | Table 1. Diagnostic Criteria of TAFRO Syndrome |
On admission, his initial body weight was 58.0 kg, blood pressure was 169/122 mm Hg, and body temperature was 38.5 °C. The patient was alert and conscious. The cervical, axillary, and inguinal lymph nodes were small, soft, and palpable. The patient was febrile, and pitting edema was noted. The patient’s abdomen is not distended.
Serological test results for hepatitis B virus (HBV), hepatitis C virus (HCV), human T-cell leukemia virus type I (HTLV-I), and HIV were negative. Serological tests for Epstein-Barr virus and cytomegalovirus revealed past infection patterns without reactivation. Polymerase chain reaction analysis of the HHV-8 sequence in the peripheral blood showed a negative result. Antinuclear antibody and rheumatoid factor test results were negative. Test results for anti-Sjogren syndrome-related antigen A and anti-Sjogren syndrome-related antigen B were negative. The antineutrophil cytoplasmic antibody (ANCA) also showed a negative result. The T-SPOT.TB assay was negative.
The initial level of hemoglobin (Hb) (10.1 g/dL, normal range: 13.7 - 16.8) and the platelet count (56,000/µL, normal range: 158,000 - 348,000) were low. Increases were noted in the leukocyte count (8,780/µL, normal range: 3,300 - 8,600) and the levels of C-reactive protein (CRP) (95.9 mg/L, normal range: < 14) and creatinine (1.70 mg/dL, normal range: 0.65 - 1.07). γ-globulin and immunoglobulin (Ig) G levels (948 mg/dL, normal range: 861 - 1,747) were within normal ranges. IgG4 levels were not elevated, and no monoclonal immunofixation bands were detected. The levels of alkaline phosphatase (ALP) (628 U/L, normal range: 106 - 322), γ-glutamyl transferase (γ-GTP) (82 U/L, normal range: 13 - 64), serum IL-6 (11.0 pg/mL, normal range: < 7.0), soluble IL-2 receptor (2,023 U/mL, normal range: 122 - 496), and serum vascular endothelial growth factor (VEGF) (5,791.4 pg/mL, normal range: 143.1 - 658.8) were elevated. Although the prothrombin time-international normalized ratio (PT-INR) and activated partial thromboplastin time (APTT) were within normal ranges, we observed increases in the levels of fibrin degradation product (FDP), measuring 43.3 µg/mL (normal range: < 5). Additionally, D-dimer level was elevated at 19.6 µg/mL (normal range: < 1.0). The data are summarized in Table 2.
Click to view | Table 2. Laboratory Data of Case 1 and Case 2 |
Treatment
After admission, the patient had a continuous fever that ranged between 38 and 39 °C, and leg pitting edema worsened. Immunotherapy was initiated after obtaining written informed consent from the patient.
Methylprednisolone pulse therapy attenuated the high fever; however, low-grade fever persisted. His CRP level remained elevated, and renal insufficiency developed (creatinine 2.46 mg/dL).
Therefore, rituximab was intravenously administered at a standard dose of 375 mg/m2, and oral prednisolone was initiated as additional maintenance therapy. Although rituximab normalized CRP and IL-6 levels, the ascites persisted and were resistant to rituximab. Subsequent administration of tocilizumab did not ameliorate ascites. Abdominal distension worsened due to ascites. Despite one infusion of tocilizumab and two doses of rituximab administered weekly, severe ascites gradually developed, and the patient’s body weight increased to 68.7 kg, marking a gain of 10.7 kg from admission.
Follow-up and outcomes
Cyclosporine was administered to the patient, which successfully resolved ascites and hepatosplenomegaly. Body weight decreased to 55.9 kg at discharge. Oral prednisolone was discontinued 4 months after the first rituximab infusion (Fig. 2).
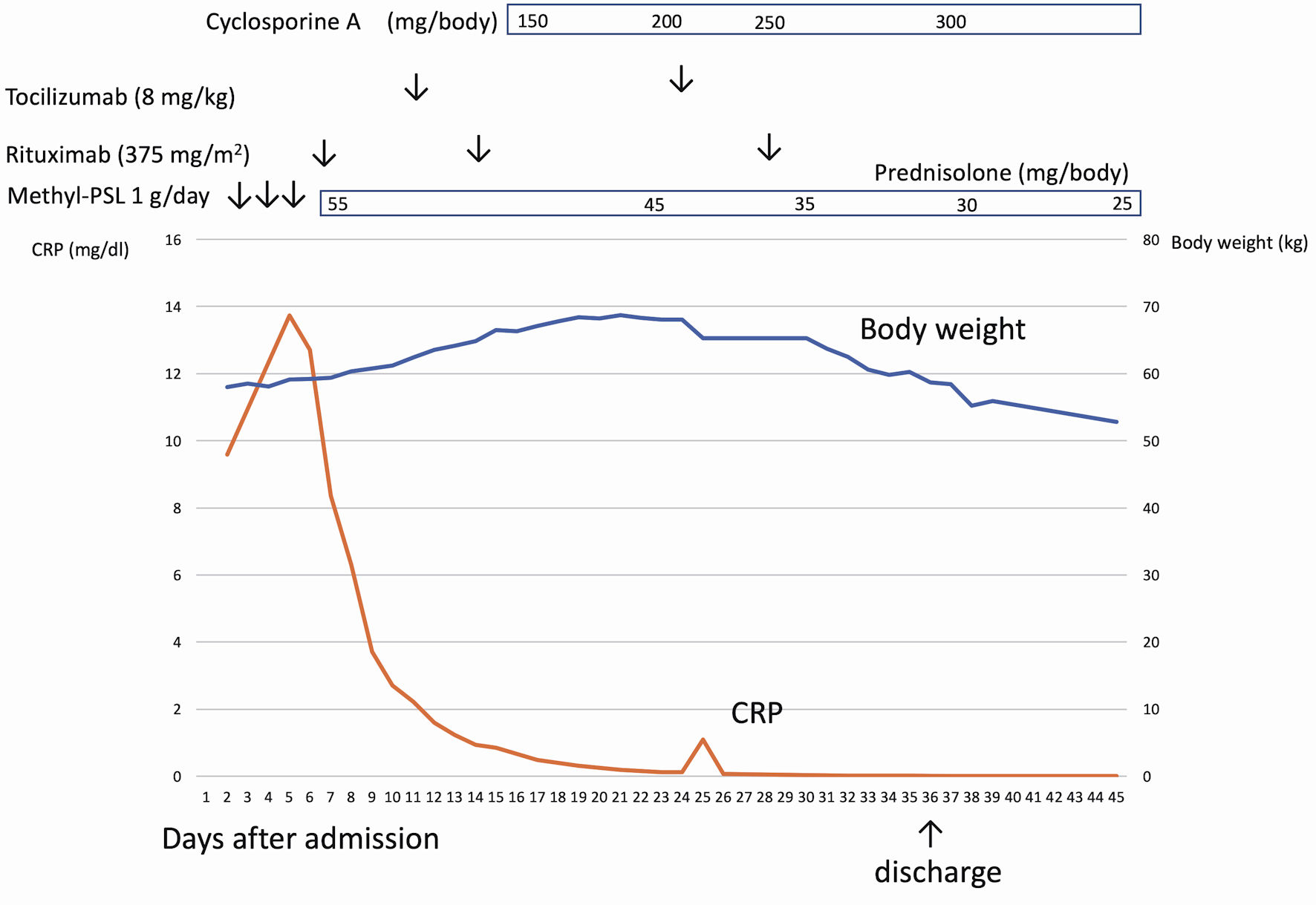 Click for large image | Figure 2. The clinical course of case 1. After admission, the patient had a continuous high fever and anasarca. After rituximab infusion, C-reactive protein (CRP) was normalized, but anasarca worsened. Cyclosporine was administered to the patient, which successfully resolved anasarca and ascites. |
Although the hypertension persisted with cyclosporine maintenance therapy for 12 months, the patient remained asymptomatic without ascites or hepatosplenomegaly (Fig. 1b).
Case 2
Investigations
A 72-year-old male patient with diabetes mellitus presented with foot and periorbital edema. Cardiac function was normal. Three months later, the patient developed severe ascites and renal dysfunction. The patient was then referred to our hospital for further examination and treatment. The renal dysfunction did not respond to diuretics or albumin infusion, and creatine levels increased. Therefore, hemodialysis was recommended. The diagnosis was not confirmed at the time when hemodialysis was initiated.
Diagnosis
Viral serological tests for HBV, HCV, HTLV-I, and HIV were negative. Serological tests for Epstein-Barr virus and cytomegalovirus revealed past infection patterns without reactivation. Polymerase chain reaction analysis of the HHV-8 sequence in the peripheral blood was negative. The antinuclear antibody test results were weakly positive. Tests for rheumatoid factor, anti-Sjogren syndrome-related antigen A, anti-Sjogren syndrome-related antigen B, ANCA, and antiplatelet antigens were negative. The T-SPOT.TB assay results were also negative.
The initial Hb level (11.0 g/dL, normal range: 13.7 - 16.8) and platelet count (67,000/µL, normal range: 158,000 - 348,000) were low. Increases were noted in the leukocyte count (5,740/µL, normal range: 3,300 - 8,600) and the levels of CRP (59.1 mg/L, normal range: < 14) and creatinine (2.74 mg/dL, normal range: 0.65 - 1.07). γ-globulin and IgG (1,187 mg/dL, normal range: 861 - 1,747) levels were within normal ranges, and the level of IgG4 was not elevated. No monoclonal immunofixation bands were observed. ALP (148 U/L, normal range: 106 - 322) and γ-GTP levels (12 U/L, normal range: 13 - 64) were not elevated. Increases were observed in serum IL-6 (55 pg/mL, normal range: < 7.0), soluble IL-2 receptor (1,629 U/mL, normal range: 122 - 496), and serum VEGF (2,566.3 pg/mL, normal range: 143.1 - 658.8) levels. PT-INR (1.3, normal range: 0.9 - 1.2) and APTT (45.0 s, normal range: 28 - 39.5) were prolonged, and FDP (30.4 µg/mL, normal range: < 5) and D-dimer (16.7 µg/mL, normal range: < 1.0) levels were elevated. The data are summarized in Table 2.
Histological examination of the bone marrow aspirate revealed myelofibrosis (Fig. 3a). An inguinal lymph node biopsy revealed atrophied germinal centers with enlarged endothelial cell nuclei (Fig. 3b). Some histological features of the lymph nodes were consistent with TAFRO-iMCD [6].
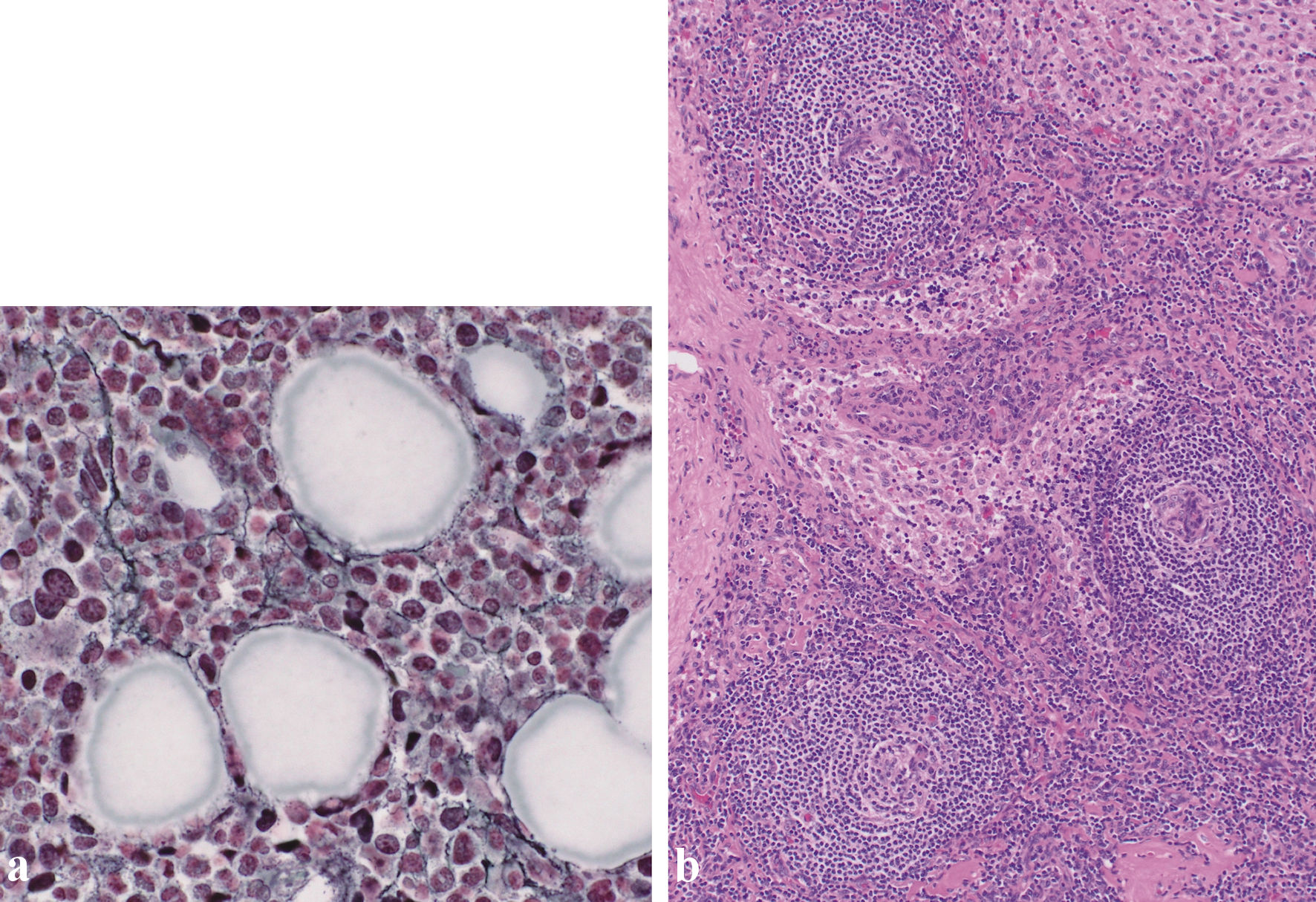 Click for large image | Figure 3. (a) Silver staining of the bone marrow revealed a network of reticulin fibers (original magnification × 400). (b) Biopsy of the right inguinal lymph node. Biopsy of the right inguinal lymph node (hematoxylin and eosin staining) revealed enlarged endothelial cell nuclei with proliferation of venules in the atrophied germinal centers. Plasma cell infiltration is observed in the intrafollicular zone (original magnification × 100). |
These findings indicated TAFRO syndrome, and the diagnosis was established based on the diagnostic criteria [7]. The diagnostic criteria are summarized in Table 1 and are compared with those of case 1.
Treatment
Immunosuppressive therapy was initiated with the written informed consent of the patient.
After rituximab infusion, CRP level decreased. Although methylprednisolone, rituximab, tocilizumab, and cyclosporine were administered in a stepwise manner, other laboratory data and clinical symptoms remained unchanged. However, the renal dysfunction did not improve after immunotherapy with hemodialysis. Therefore, cyclophosphamide pulse therapy was initiated but was ineffective.
Follow-up and outcomes
The patient developed herpes zoster, experienced a decline in consciousness, and eventually passed away (Fig. 4).
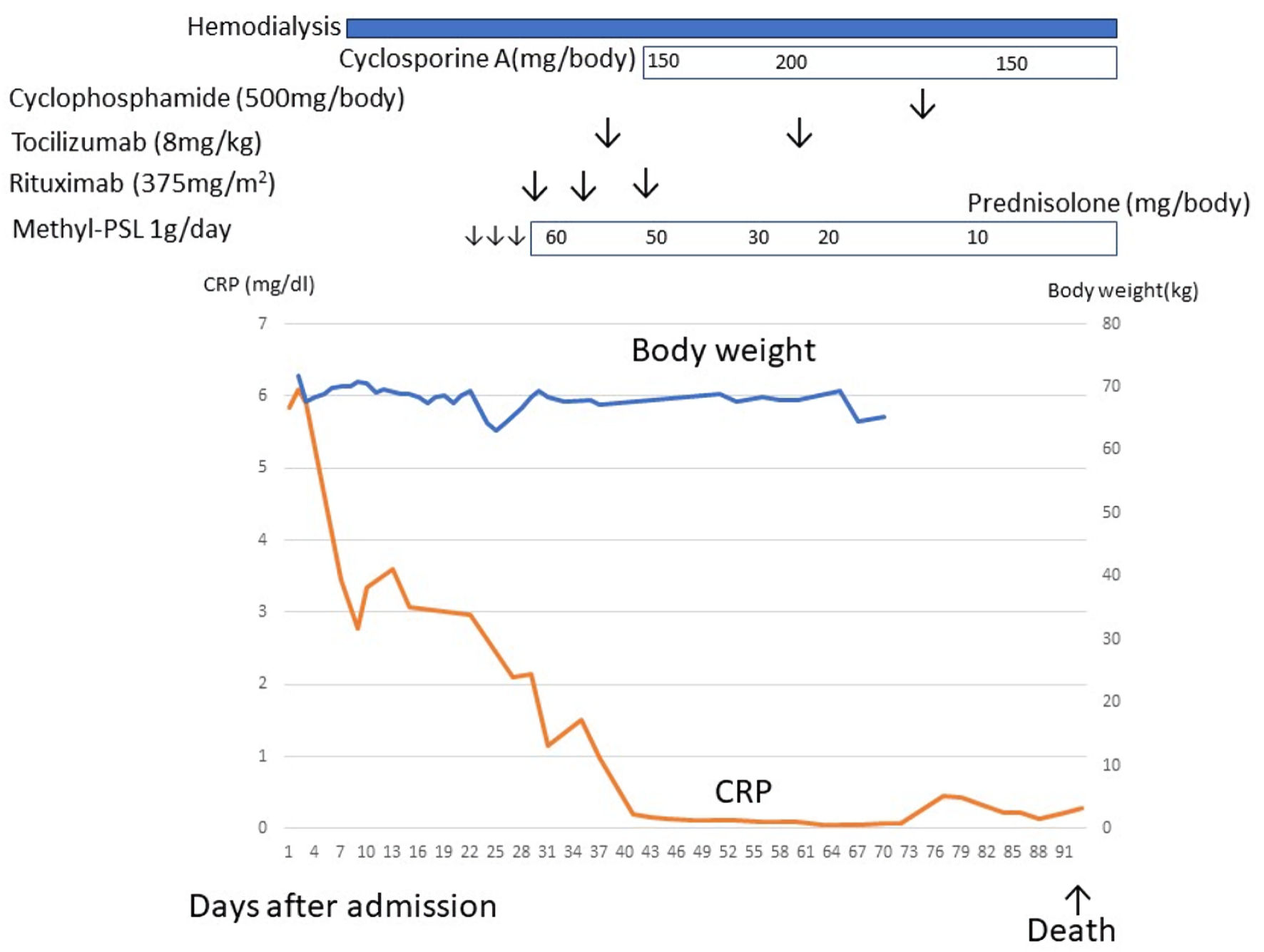 Click for large image | Figure 4. The clinical course of case 2. After admission, the patient developed severe ascites and renal dysfunction. Renal dysfunction did not respond to diuretics, and hemodialysis was indicated. After a diagnosis of TAFRO syndrome, methylprednisolone, rituximab, tocilizumab, and cyclosporine, cyclophosphamide therapy was initiated but ineffective. This patient died due to herpes zoster encephalitis. CRP: C-reactive protein. |
The autopsy revealed severe ascites (9 L), bilateral bacterial pneumonia, myelofibrosis, lymphadenopathy, congestive spleen, and atherosclerosis. Histopathological examination of the kidneys revealed early diabetic nephritis without severe damage.
Herpes zoster encephalitis was diagnosed based on a positive cerebrospinal fluid polymerase chain reaction. the causes of death were attributed to herpes zoster encephalitis and bacterial pneumonia, likely resulting from overimmunosuppression.
| Discussion | ▴Top |
We treated two patients with TAFRO using a new stepwise immunotherapy consisting of rituximab and cyclosporine. One patient achieved remission, whereas the other died.
TAFRO syndrome was originally described by Takai et al in three Japanese patients; it was initially described as a new syndrome based on the combination of typical symptoms and laboratory findings (fever, anasarca, hepatosplenomegaly, lymphadenopathy, thrombocytopenia, and reticulin fibrosis in bone marrow) in 2010 [13].
Lymphadenopathy is a common feature of TAFRO syndrome, but mild lymph node biopsy may not be feasible in certain cases. In cases of TAFRO syndrome in which lymph node biopsy is possible, the pathology of the enlarged lymph nodes resembles CD (TAFRO-iMCD) [6]. In some cases of TAFRO syndrome, lymph node biopsy was clinically impossible owing to general conditions (such as bleeding tendency); thus, a pathological diagnosis was not made. Cases of TAFRO syndrome were classified as TAFRO syndrome without proven iMCD (TAFRO-w/op-iMCD). No significant differences in the clinical, laboratory, or prognostic features were noted between the TAFRO-iMCD and TAFRO-w/op-iMCD [8]. However, the clinical symptoms and laboratory data differ between TAFRO syndrome (TAFRO-iMCD and TAFRO-w/op-iMCD) and typical iMCD (iMCD-NOS). The main symptoms, such as anemia, lymphadenopathy, and kidney damage, are common in these two diseases, although the speed of progression differs. TAFRO syndrome is characterized by rapid, acute progression over a short period, while iMCD typically follows an indolent course, developing over a more extended duration. Normal or hypogammaglobulinemia and high ALP levels are indicative of the TAFRO syndrome, whereas hypergammaglobulinemia is suggestive of iMCD.
CD has been reported as a mediastinal tumor with an unusual pathology [2] and no obvious symptoms or laboratory findings. MCD is characterized by nonspecific symptoms of inflammation, such as generalized lymphadenopathy, fever, and night sweats. Patients die from infectious diseases and/or multi-organ failure due to MCD in severe cases [1]. The histopathology of CD was originally divided into three categories: hyaline vasculature, plasma cell, and mixed. In summary, CD is typically diagnosed based on lymph node histopathology, whereas TAFRO syndrome is diagnosed based on clinical symptoms and laboratory findings.
Hence, many diseases with varying etiologies and symptoms are categorized as CD, including iMCD-NOS, TAFRO-iMCD, HHV-8 associated MCD, and POEMS syndrome-associated MCD, primarily due to similar lymph node histopathology [1].
It remains uncertain whether TAFRO syndrome should be regarded as a subtype of iMCD or as an independent disease unrelated to CD. Nevertheless, the clinical courses of patients with iMCD and those with TAFRO differ significantly. Consequently, there is a clear need for clinical and therapeutic distinctions [9].
In case 1, the infusion of rituximab normalized the levels of IL-6 and inflammatory parameters (such as CRP), whereas ascites and edema worsened. The ascites was resolved by the administration of cyclosporine. Tocilizumab was ineffective as second-line treatment for TAFRO syndrome in case 1. However, the role of tocilizumab in TAFRO syndrome treatment remains unclear.
In case 2, stepwise immunotherapy was completely ineffective, and the patient died of herpes encephalitis and bacterial pneumonia due to overimmunosuppression.
The reason for the difference in treatment effectiveness between cases 1 and 2 remains unclear. However, it is worth noting that by the time immunotherapy was initiated in case 2, the disease had progressed to a stage requiring dialysis, which might have negatively influenced its effectiveness. The histopathology of the kidneys did not show features of TAFRO syndrome, such as membranoproliferative glomerulonephritis, and TMA-like histological findings [14]. Furthermore, severe kidney damage was not detected; therefore, renal failure may have developed owing to a cytokine storm and dehydration. The pathological autopsy in case 2 did not show any other malignancies or immune disorders, and the diagnosis of TAFRO syndrome was not problematic.
The treatment guidelines for iMCD recommend anti-IL-6 therapy (tocilizumab and siltuximab) and/or rituximab. Anti-IL-6 therapy blocks inflammation caused by hypercytokinesis and is not curative. Therefore, the therapeutic effects of anti-IL-6 therapy disappear within a short period and require repeated administration of tocilizumab or siltuximab. Prospective studies have demonstrated the efficacy of rituximab for HHV-8-associated CD, and its efficacy has been confirmed in clinical studies [15-17]. In contrast, the efficacy of rituximab in iMCD has only been reported in small case series [18, 19]. Therefore, anti-IL-6 therapy is generally recommended for severe iMCD, whereas rituximab is only recommended for mild iMCD cases [11].
The effects of rituximab on iMCD are inferior to those of anti-IL-6 therapy. However, tocilizumab was previously reported to be effective for TAFRO syndrome [20] but was associated with a poorer prognosis than rituximab as the initial treatment [12]. Rituximab may have a more pronounced effect on TAFRO syndrome (TAFRO-iMCD), as opposed to iMCD-NOS.
Many effective agents have been reported for the treatment of the TAFRO syndrome, including rituximab, tocilizumab, cyclosporine [9], and cyclophosphamide [21].
Rituximab is used in anti-B-cell therapy, tocilizumab in anti-IL-6 therapy [6], and cyclosporine and/or cyclophosphamide in anti-T-cell therapy [22, 23]. These agents are generally used to treat the TAFRO syndrome; however, a systematic treatment algorithm has not yet been established. Currently, the treatment of TAFRO syndrome has primarily involved the use of single agents, with limited attempts at combination therapy. In the future, combination therapy with immunotherapeutic agents may be effective.
Most cases of iMCD-NOS may be controlled by anti-IL-6 therapy (tocilizumab and siltuximab) and/or rituximab [11], whereas TAFRO syndrome may require additional suppression of T-cell systems. Recently, mTOR activation related to iMCD and TAFRO syndrome has been reported [24, 25]. Based on these findings, sirolimus was shown to be effective against tocilizumab-refractory iMCD [26]; and based on these findings, a clinical study on sirolimus for iMCD has been initiated [27]. However, the prognosis of TAFRO syndrome (TAFRO-iMCD) initially treated with cyclosporine (anti-T-cell therapy) is poor [12]. Consequently, it is recommended that after the initial treatment with rituximab and the confirmation of improvements in clinical symptoms and laboratory data (including IL-6 levels and inflammatory parameters like CRP), initiating cyclosporine maintenance therapy is advisable. Although there are still a few cases under consideration, rituximab with cyclosporine therapy may be the preferred first-line therapy for steroid-resistant TAFRO syndrome.
In conclusion, treatment guidelines for iMCD recommend anti-IL-6 therapy and/or rituximab, which control most cases of iMCD. However, different treatments may be required for the TAFRO syndrome. We consider the combination of rituximab induction therapy and cyclosporine maintenance therapy to be effective for TAFRO syndrome if initiated at an early stage.
Learning points
The treatment guidelines for iMCD recommend anti-IL-6 therapy and/or rituximab, which controls most cases of iMCD. However, different treatments may be required for the TAFRO syndrome. We consider the combination of rituximab induction therapy and cyclosporine maintenance therapy to be effective for TAFRO syndrome if initiated at an early stage.
Acknowledgments
The authors thank the patients, their families, and the Castleman Disease Collaborative Network.
Financial Disclosure
None to declare.
Conflict of Interest
The authors have no conflict of interest to declare.
Informed Consent
Informed consent was obtained in the form of an opt-out on website.
Author Contributions
M. Ide, as chief director of the Hematology Department, selected the treatment plan. TY provided treatment, including dialysis, as the doctor in charge. M. Ishikawa described pathological findings on the specimen as a pathologist. KK provided treatment as a doctor in charge. All the authors have read and approved the final version of the manuscript.
Data Availability
All clinical and laboratory data were examined at the Takamatsu Red Cross Hospital or extracted from letters referring to the Takamatsu Red Cross Hospital.
Abbreviations
ANCA: antineutrophil cytoplasmic antibody; APTT: activated partial thromboplastin time; CD: Castleman disease; CRP: C-reactive protein; CT: computed tomography; FDP: fibrin degradation product; Hb: hemoglobin; HBV: hepatitis B virus; HCV: hepatitis C virus; HHV-8: human herpes virus 8; HIV: human immunodeficiency virus; HTLV-I: human T-cell leukemia virus type I; Ig: immunoglobulin; IL: interleukin; iMCD: idiopathic multicentric Castleman disease; iMCD-NOS: idiopathic multicentric Castleman disease-not otherwise specified; MCD: multicentric Castleman disease; PT-INR: prothrombin time-international normalized ratio; UCD: unicentric Castleman disease; VEGF: vascular endothelial growth factor
| References | ▴Top |
- Dispenzieri A, Fajgenbaum DC. Overview of Castleman disease. Blood. 2020;135(16):1353-1364.
doi pubmed - Castleman B, Iverson L, Menendez VP. Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer. 1956;9(4):822-830.
doi pubmed - Gaba AR, Stein RS, Sweet DL, Variakojis D. Multicentric giant lymph node hyperplasia. Am J Clin Pathol. 1978;69(1):86-90.
doi pubmed - Frizzera G, Banks PM, Massarelli G, Rosai J. A systemic lymphoproliferative disorder with morphologic features of Castleman's disease. Pathological findings in 15 patients. Am J Surg Pathol. 1983;7(3):211-231.
doi pubmed - Fajgenbaum DC, Uldrick TS, Bagg A, Frank D, Wu D, Srkalovic G, Simpson D, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood. 2017;129(12):1646-1657.
doi pubmed pmc - Iwaki N, Fajgenbaum DC, Nabel CS, Gion Y, Kondo E, Kawano M, Masunari T, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J Hematol. 2016;91(2):220-226.
doi pubmed - Masaki Y, Kawabata H, Takai K, Tsukamoto N, Fujimoto S, Ishigaki Y, Kurose N, et al. 2019 Updated diagnostic criteria and disease severity classification for TAFRO syndrome. Int J Hematol. 2020;111(1):155-158.
doi pubmed - Fujimoto S, Sakai T, Kawabata H, Kurose N, Yamada S, Takai K, Aoki S, et al. Is TAFRO syndrome a subtype of idiopathic multicentric Castleman disease? Am J Hematol. 2019;94(9):975-983.
doi pubmed - Masaki Y, Arita K, Sakai T, Takai K, Aoki S, Kawabata H. Castleman disease and TAFRO syndrome. Ann Hematol. 2022;101(3):485-490.
doi pubmed pmc - van Rhee F, Oksenhendler E, Srkalovic G, Voorhees P, Lim M, Dispenzieri A, Ide M, et al. International evidence-based consensus diagnostic and treatment guidelines for unicentric Castleman disease. Blood Adv. 2020;4(23):6039-6050.
doi pubmed pmc - van Rhee F, Voorhees P, Dispenzieri A, Fossa A, Srkalovic G, Ide M, Munshi N, et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood. 2018;132(20):2115-2124.
doi pubmed pmc - Fujimoto S, Kawabata H, Sakai T, Yanagisawa H, Nishikori M, Nara K, Ohara S, et al. Optimal treatments for TAFRO syndrome: a retrospective surveillance study in Japan. Int J Hematol. 2021;113(1):73-80.
doi pubmed - Takai K, Nikkuni K, Shibuya H, Hashidate H. [Thrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegaly]. Rinsho Ketsueki. 2010;51(5):320-325.
pubmed - Nagayama Y, Yamano M, Yagame M, Nariyama T, Takahashi M, Kawamoto M, Matsui K. TAFRO syndrome as a cause of glomerular microangiopathy: a case report and literature review. BMC Nephrol. 2019;20(1):375.
doi pubmed pmc - Bower M, Powles T, Williams S, Davis TN, Atkins M, Montoto S, Orkin C, et al. Brief communication: rituximab in HIV-associated multicentric Castleman disease. Ann Intern Med. 2007;147(12):836-839.
doi pubmed - Gerard L, Berezne A, Galicier L, Meignin V, Obadia M, De Castro N, Jacomet C, et al. Prospective study of rituximab in chemotherapy-dependent human immunodeficiency virus associated multicentric Castleman's disease: ANRS 117 CastlemaB Trial. J Clin Oncol. 2007;25(22):3350-3356.
doi pubmed - Uldrick TS, Polizzotto MN, Aleman K, Wyvill KM, Marshall V, Whitby D, Wang V, et al. Rituximab plus liposomal doxorubicin in HIV-infected patients with KSHV-associated multicentric Castleman disease. Blood. 2014;124(24):3544-3552.
doi pubmed pmc - Ide M, Kawachi Y, Izumi Y, Kasagi K, Ogino T. Long-term remission in HIV-negative patients with multicentric Castleman's disease using rituximab. Eur J Haematol. 2006;76(2):119-123.
doi pubmed - Ide M, Ohnishi H, Fukumoto T, Ohno H, Yokoyama T. Re-evaluation of rituximab therapy for idiopathic Castleman disease: Retrospective study from single-center experience. Eur J Haematol. 2022;108(4):354-355.
doi pubmed - Akiyama M, Kaneko Y, Takeuchi T. Tocilizumab for the treatment of TAFRO syndrome: a systematic literature review. Ann Hematol. 2020;99(11):2463-2475.
doi pubmed - Kikuchi T, Shimizu T, Toyama T, Abe R, Okamoto S. Successful treatment of TAFRO syndrome with tocilizumab, prednisone, and cyclophosphamide. Intern Med. 2017;56(16):2205-2211.
doi pubmed pmc - Hughes E, Scurr M, Campbell E, Jones E, Godkin A, Gallimore A. T-cell modulation by cyclophosphamide for tumour therapy. Immunology. 2018;154(1):62-68.
doi pubmed pmc - Liddicoat AM, Lavelle EC. Modulation of innate immunity by cyclosporine A. Biochem Pharmacol. 2019;163:472-480.
doi pubmed - Phillips AD, Kakkis JJ, Tsao PY, Pierson SK, Fajgenbaum DC. Increased mTORC2 pathway activation in lymph nodes of iMCD-TAFRO. J Cell Mol Med. 2022;26(11):3147-3152.
doi pubmed pmc - Arenas DJ, Floess K, Kobrin D, Pai RL, Srkalovic MB, Tamakloe MA, Rasheed R, et al. Increased mTOR activation in idiopathic multicentric Castleman disease. Blood. 2020;135(19):1673-1684.
doi pubmed pmc - Sumiyoshi R, Koga T, Furukawa K, Umeda M, Yamamoto K, Mori R, Kawakami A. A case of tocilizumab-refractory idiopathic multicentric Castleman's disease successfully treated with sirolimus. Clin Immunol. 2021;233:108887.
doi pubmed - Koga T, Hagimori N, Takemori S, Morimoto S, Sumiyoshi R, Shimizu T, Hosogaya N, et al. Randomized, double-blind, placebo-controlled, parallel-group trial of sirolimus for tocilizumab-resistant idiopathic multicentric Castleman disease: Study protocol for clinical trial. Medicine (Baltimore). 2020;99(30):e20710.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.