

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 3, Number 4, August 2012, pages 219-222
Hemophagocytic Syndrome In Association With Systemic Lupus Erythematosus and Rheumatoid Arthritis
Mohammad Esmadia, c, Muhammad O. Khokhara, Dina S. Ahmada, Donald C. Dollb
aDepartment of Internal Medicine, University of Missouri School of Medicine, Columbia, MO, USA
bDivision of Hematology and Medical Oncology, Department of Internal Medicine, University of Missouri School of Medicine, Columbia, MO, USA
cCorresponding author: Mohammad Esmadi, One Hospital Dr., DC043.00 Health Sciences Center, Columbia, MO 65212, USA
Manuscript accepted for publication March 13, 2012
Short title: HPS in Association With SLE and RA
doi: https://doi.org/10.4021/jmc589w
| Abstract | ▴Top |
Hemophagocytic syndrome (HPS) is a potentially life-threatening complication of rheumatic disorders characterized by the infiltration of morphologically benign hemophagocytic macrophages in the bone marrow and in various other organs. It is known to be associated with several conditions, such as viral or bacterial infection, malignancies, and autoimmune diseases such as systemic lupus erythematosus (SLE) and adult onset Still’s disease and less commonly with rheumatoid arthritis(RA). We report a case of a 59-year-old lady known to have SLE and RA who developed worsening anemia, thrombocytopenia, coagulopathy, renal failure and was diagnosed with hemophagocytic syndrome. In spite of aggressive supportive management, intravenous steroids and immunoglobulins, the patient eventually died due to intracranial bleeding. Although cases of HPS have been previously described in association with SLE or RA, this is the first case of HPS to our knowledge to be diagnosed in a patient who had both RA and SLE.
Keywords: Hemophagocytic syndrome; Systemic lupus erythematosus; Rheumatoid arthritis; Lymphohistiocytoses
| Introduction | ▴Top |
Hemophagocytic syndrome (HPS) is an entity characterized by the activation of macrophages and/or histiocytes with prominent hemophagocytosis in bone marrow and other reticuloendothelial systems [1]. HPS comprises primary (hereditary) and reactive forms. Primary HPS denotes the presence of an underlying genetic disorder and is observed mostly in infants. Reactive HPS occurs in situations such as infections, malignant lymphomas, a variety of autoimmune diseases including lupus erythematosus, rheumatoid arthritis, Still's disease, polyarteritis nodosa, mixed connective tissue disease, pulmonary sarcoidosis, systemic sclerosis, and Sjogren's syndrome, and certain drugs [2-4]. Clinical features of HPS include fever, cytopenia, liver enzyme elevation, hepatosplenomegaly, lymphadenopathy, elevated ferritin levels, hypertriglyceremia and coagulopathy [5].
| Case Report | ▴Top |
We describe a 59-year-old white female who is known to have systemic lupus erythematousus (SLE) and rheumatoid arthritis (RA), on methotrexate 2.5 mg every week, and prednisone 5 mg daily who presented with nausea, vomiting, diarrhea and fatigue over a period of one week. Her vital signs were stable and her physical examination was unremarkable. On admission, her creatinine was 4.4 mg/dL, hemoglobin 14.4 gm/dl, platelets 110,000/mcL, white cell count 3210/mcL, total bilirubin 0.2 mg/dL, AST 47 unit/L, ALT 46 unit/L, alkaline phosphatase 193 unit/L, total protein 7.7 g/dL, albumin 3.9 g/dL, lactate dehydrogenase (LDH) 491 unit/L, INR 0.9 and partial thromboplastin time (PTT) 58 seconds.
Soon after admission, however, she started to spike fevers and her hemoglobin dropped from 14.4 gm/dl to 10.4 gm/dL, and platelets dropped to 53,000/mcL. Her PTT increased to 70 seconds. Peripheral smear showed normocytic cells with burr cells and thrombocytopenia, but no shistocytes. PTT mixing study corrected partially with PTT remaining elevated at 39.7 seconds. Factor V Assay was 97%, factor IX 67%, factor X 100%, factor VII 182% and factor VIII-C 236%. Thrombin time was 60 seconds. She received prophylactic subcutaneous heparin during the first 3 days of her hospitalization, but platelet factor 4 (PF4) antibody was normal. Her fibrinogen had always been greater than 190 mg/dL. The patient also tested negative for HIV, EBV and viral hepatitis.
Bone marrow biopsy was done on day 5. After the bone marrow biopsy the patient began to bleed from the biopsy site along with mucosal and subcutaneous bleeding. Her PTT at this point was around 100 seconds. She became hypotensive and was transferred to the ICU and started on broad spectrum antibiotics. All cultures were negative. Due to worsening anemia, she required 10 units of blood along with multiple units of fresh frozen plasma and cryoprecipitate. Her platelets continued to drop down to 8000/mcL.
On day number 7, the bone marrow biopsy result came back as 15% plasma cells with hemophagocytic macrophages (Fig. 1), raising the suspicion for HPS. Moreover, ferritin level was 17289 ng/mL and triglycerides level was elevated at 657 mg/dL. The patient was started on Methylprednisolone 1000 mg/day and intravenous immunoglobulins (IVIG) 1g/kg/day. Her kidney function continued to deteriorate and required dialysis eventually. Her level of consciousness also started to deteriorate. Her platelets and hemoglobin continued to drop and required repeated transfusions of platelets and blood. IVIG was stopped on day 9. On day number 10, her triglycerides dropped to 525 mg/dL and ferritin dropped to 7,791 ng/mL. On day 11, she had a negative work up for ANCA and anti-cardiolipin antibodies. ADAMTS13 activity was decreased at 58%.
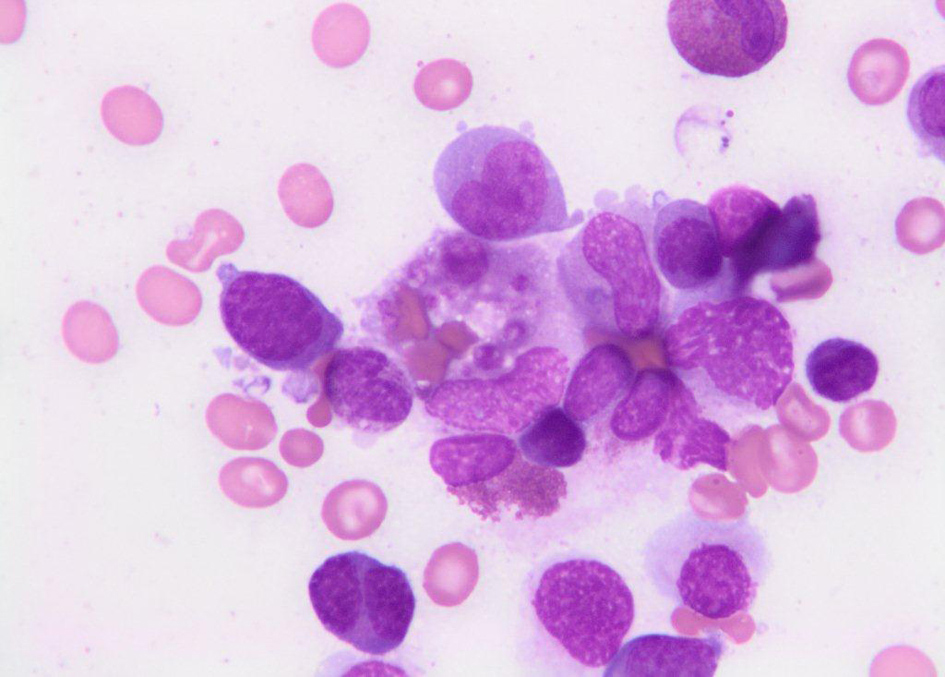 Click for large image | Figure 1. Bone marrow biopsy showing hemophagocytic macrophages. |
On day 12, the patient had even more worsening of her level of consciousness. MRI indicated diffuse cortical necrosis/hemorrhage. Given, the MRI findings and the worsening renal failure, coagulopathy, anemia and thrombocytopenia, the decision was to defer any further aggressive supportive measures and she eventually died.
| Discussion | ▴Top |
The widely used criteria for diagnosing MAS are the hemophagocytic lymphohistiocytosis (HLH) criteria, which include fever, splenomegaly, cytopenias affecting at least two of three lineages in the peripheral blood, hypertriglyceridemia and/or hypofibrinogenemia, hemophagocytosis in bone marrow, spleen or lymph nodes, low or absent natural killer cell activity, hyperferritinemia, and high levels of sIL-2r (Table 1). Altogether, five of the eight criteria must be fulfilled; patients with a molecular diagnosis consistent with HLH do not necessarily need to fulfill the diagnostic criteria [6-9].
 Click to view | Table 1. Revised Diagnostic Guidelines for Hemophagocytic Lymphohistiocytosis (HLH) |
In a study done by Fukaya et al, the prevalence of HPS among inpatients with autoimmune diseases was 3.0%. HPS was most prevalent in patients with adult onset Still’s disease, followed by Sjogren's Syndrome and SLE. At the time when the diagnosis of HPS was made, fever was observed in 87% of the patients. Neuropsychiatric manifestations were noted in 30%. Leukopenia, anemia or thrombocytopenia was found in 87, 83 or 87%, respectively. CRP was elevated in 83% of the patients. Liver enzyme elevation (aspartate aminotransferase or LDH), more than two times the upper normal limit, was observed in 60%. Median ferritin level was 980 ng/ml. Factors related with mortality were age over 50 years, the presence of infection, leucocyte count < 0.5 x 109/L, platelet count < 50 x 109/L and CRP level < 50 mg/L at the onset of HPS. [5] Current treatment strategy include induction therapy over an eight-week period with dexamethasone, etoposide (VP-16), and intrathecal methotrexate, followed by cyclosporine (trough levels of 200 µ/L) started at week 9, along with pulses of dexamethasone and etoposide for up to one year [6].
Our patient had fever, bicytopenia, hepatic dysfunction, hypertriglyceridemia, hyperferritinemia and hemophagocytic macrophages on bone marrow aspirate. The ADAMS13 of 58 % is non-specific and could be explained by the liver disease the patient had. Plasma cells in the bone marrow biopsy are related to the connective tissue disease the patient had rather than multiple myeloma. HPS might have been precipitated by an undetected gastroenteritis or the connective tissue diseases. The drop in ferritin and triglycerides indicates response to Methylprednisolone and IVIG. Although cases of HPS were previously described to be associated with SLE or RA, this is the first case of HPS to our knowledge to be reported in a patient who had both RA and SLE.
Acknowledgments
Work done at University of Missouri School of Medicine, Columbia, MO.
Conflict of Interest
No conflict of interest.
Grant Support
No Funding.
| References | ▴Top |
- Imashuku S. Differential diagnosis of haemophagocytic syndrome: underlying disorders and selection of the most effective treatment. Int J Hematol. 1997;66:135–51.
- Kumakura S, Ishikura H, Umegae N, Yamagata S, Kobayashi S. Autoimmune-associated hemophagocytic syndrome. Am J Med. 1997;102(1):113-115.
pubmed doi - Dhote R, Simon J, Papo T, Detournay B, Sailler L, Andre MH, Dupond JL, et al. Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum. 2003;49(5):633-639.
pubmed doi - Arlet JB, Le TH, Marinho A, Amoura Z, Wechsler B, Papo T, Piette JC. Reactive haemophagocytic syndrome in adult-onset Still's disease: a report of six patients and a review of the literature. Ann Rheum Dis. 2006;65(12):1596-1601.
pubmed doi - Fukaya S, Yasuda S, Hashimoto T, Oku K, Kataoka H, Horita T, Atsumi T, et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30 cases. Rheumatology (Oxford). 2008;47(11):1686-1691.
pubmed doi - Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991;18(1):29-33.
pubmed - Henter J-I, Tondini C, Pritchard J. Histiocytic syndromes. Crit Rev Oncol Hematol. 2004;50:157−74.
- Kelly A, Ramanan AV. Recognition and management of macrophage activation syndrome in juvenile arthritis. Curr Opin Rheumatol. 2007;19(5):477-481.
pubmed doi - Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131.
pubmed doi
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.