

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 8, Number 12, December 2017, pages 378-380
Not Just Another Aspiration Pneumonia: Oculopharyngeal Muscular Dystrophy
Simant Singh Thapaa, e, Abhijai Singhb, Jeffrey Scottc, Anthony Izzod
aDepartment of Internal Medicine, Saint Vincent Hospital, University of Massachusetts Medical School, Worcester, MA 01608, USA
bDepartment of Internal Medicine, Saint Vincent Hospital, Worcester, MA 01608, USA
cDepartment of Pulmonary and Critical Care, Saint Vincent Hospital, Worcester, MA 01608, USA
dDepartment of Neurology, Saint Vincent Hospital, University Of Massachusetts Medical School, Worcester, MA 01608, USA
eCorresponding Author: Simant Singh Thapa, Department of Internal Medicine, Saint Vincent Hospital, University of Massachusetts Medical School, Worcester, MA 01608, USA
Manuscript submitted September 23, 2017, accepted October 12, 2017
Short title: Oculopharyngeal Muscular Dystrophy
doi: https://doi.org/10.14740/jmc2928w
| Abstract | ▴Top |
Oculopharyngeal muscular dystrophy (OPMD) is a rare genetic myopathy of adulthood onset which presents as late-onset ptosis, dysphagia and family history of ptosis and dysphagia. It occurs due to the poly(A) binding nuclear protein 1 (PABPN1) gene mutation. A 75-year-old male presented with complaints of fever, cough and worsening dysphagia. His father and paternal grandfather both had a history of ptosis and dysphagia. He was febrile, tachypneic and bilateral ptosis was noticed. The chest CT revealed consolidation. A modified barium swallow showed frank tracheal aspiration. An acetylcholine receptor antibody test was negative. An electromyogram and nerve conduction study revealed a generalized myopathic process with the absence of myotonia. With a history of adult-onset bilateral ptosis and progressive dysphagia along with the positive family history of ptosis and dysphagia, there was a strong suspicion for OPMD. Genetic testing was performed, which was positive as it detected GCG expansion in the PABPN1 gene, which is associated with OPMD and thus the diagnosis of OPMD was established. Malnutrition and recurrent aspiration are potential and harmful complications that reduce the life expectancy of these patients.
Keywords: Hereditary ptosis; Progressive dysphagia; OPMD
| Introduction | ▴Top |
Oculopharyngeal muscular dystrophy (OPMD) is a rare, late-onset, inherited myopathic disorder which presents as progressive ptosis, worsening dysphagia, and proximal muscle weakness, in a variable combination and severity. These symptoms usually start around fourth to the sixth decade of life and thus OPMD can be easily overlooked [1, 2]. Here we present a case of OPMD, who was initially admitted as aspiration pneumonia and later on was found out to have underlying OPMD.
| Case Report | ▴Top |
A 75-year-old male presented to the hospital with a complaint of fever, cough of 1 week duration and worsening of 2 years of chronic dysphagia. His cough was productive with yellowish phlegm which was not blood mixed. He was having a persistent fever and started noticing difficulty breathing even on rest for which he presented to the emergency department. His dysphagia started around 2 years ago which progressively got worse with difficulty swallowing mainly solid meals. He had modified his swallowing technique by himself to avoid choking and he had no previous history of aspiration. His past medical history was adult-onset bilateral ptosis that was first noticed when he was around mid-50s. His ptosis had also progressed significantly over the years and he was offered blepharoplasty by an ophthalmologist in past which he had refused. His father and paternal grandfather also had a history of ptosis and dysphagia. His father had deceased from a complication of aspiration pneumonia. On physical examination, he was febrile and tachypneic with bilateral rhonchi on chest auscultation. Bilateral ptosis was present (Fig. 1). Cranial nerve examination was intact. The motor exam revealed 3/5 weakness of the bilateral infraspinatus and other shoulder girdle muscles. Distal arm and forearm muscle groups including biceps, triceps, brachioradialis, wrist flexors and extensors and intrinsic hand muscles were 5/5. Muscle strength in the bilateral lower extremities was 5/5 in all muscle groups tested. The sensation was intact throughout. Ataxia was absent. He had an elevated WBC of 19,000 and a chest CT revealed consolidation in the left upper and lower lobes and in the right lower lobe posteriorly (Fig. 2). A modified barium swallow showed frank tracheal aspiration (Fig. 3). An acetylcholine receptor antibody test was negative. An electromyogram and nerve conduction study revealed a generalized myopathic process with the absence of myotonia. With a history of adult-onset bilateral ptosis and progressive dysphagia with aspiration complication along with the positive family history of ptosis and dysphagia, there was a strong suspicion for OPMD. Genetic testing was performed, which was positive as it detected GCG expansion, with expanded allele size of 13 repeats, in the poly(A) binding nuclear protein 1 (PABPN1) gene, which is associated with OPMD and thus the diagnosis of OPMD was established. He was treated with antibiotics for aspiration pneumonia along with supportive management. Initially, the patient required high flow oxygen but with resolution of pneumonia his respiratory status improved and he was not requiring any oxygen support at the time of discharge. He was offered surgical corrective measures to help him with his ptosis and dysphagia but he declined any surgical interventions. At the time of discharge, the patient was offered a percutaneous endoscopic gastrostomy (PEG) tube placement to help him maintain proper nutritional status due to underlying severe dysphagia, which he accepted. Genetic counseling for his family members was recommended after his diagnosis.
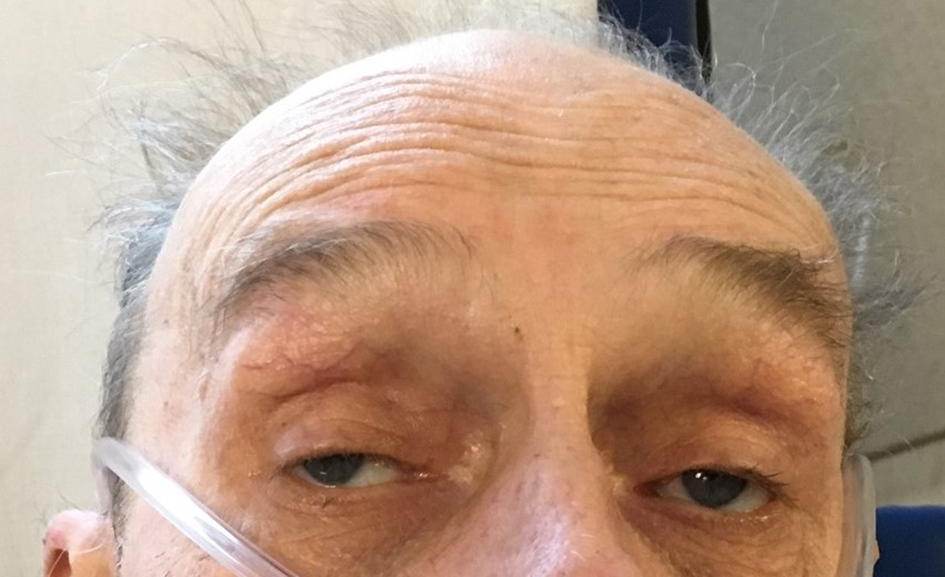 Click for large image | Figure 1. Bilateral ptosis with tendency to contract the forehead muscle to raise the eyelids. |
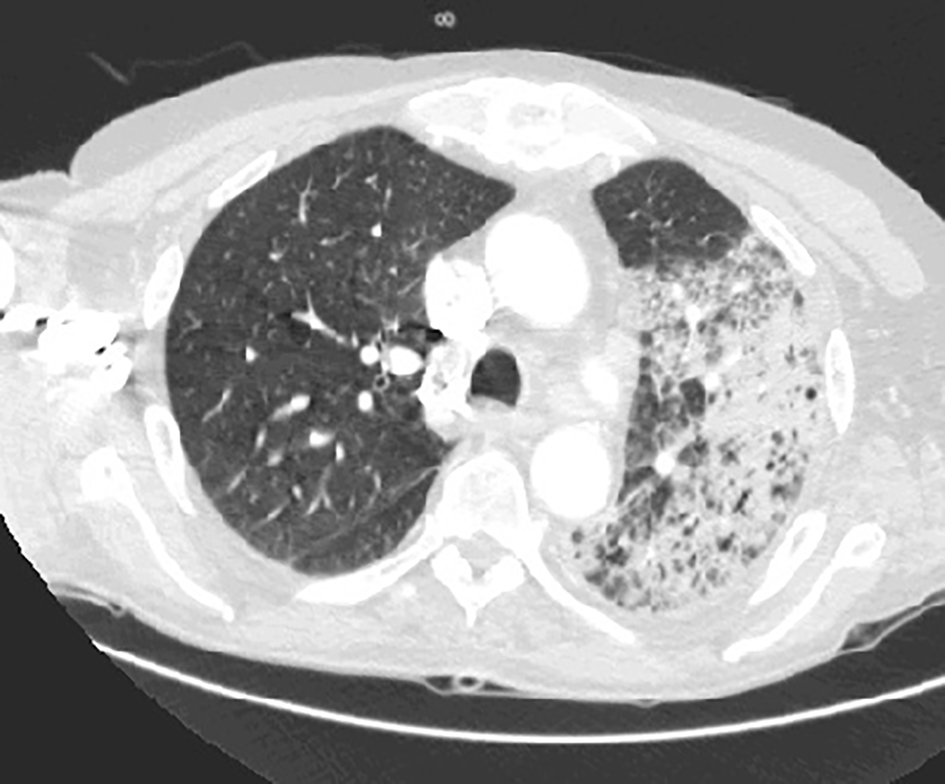 Click for large image | Figure 2. Chest CT showing consolidation, predominantly in the left lung. |
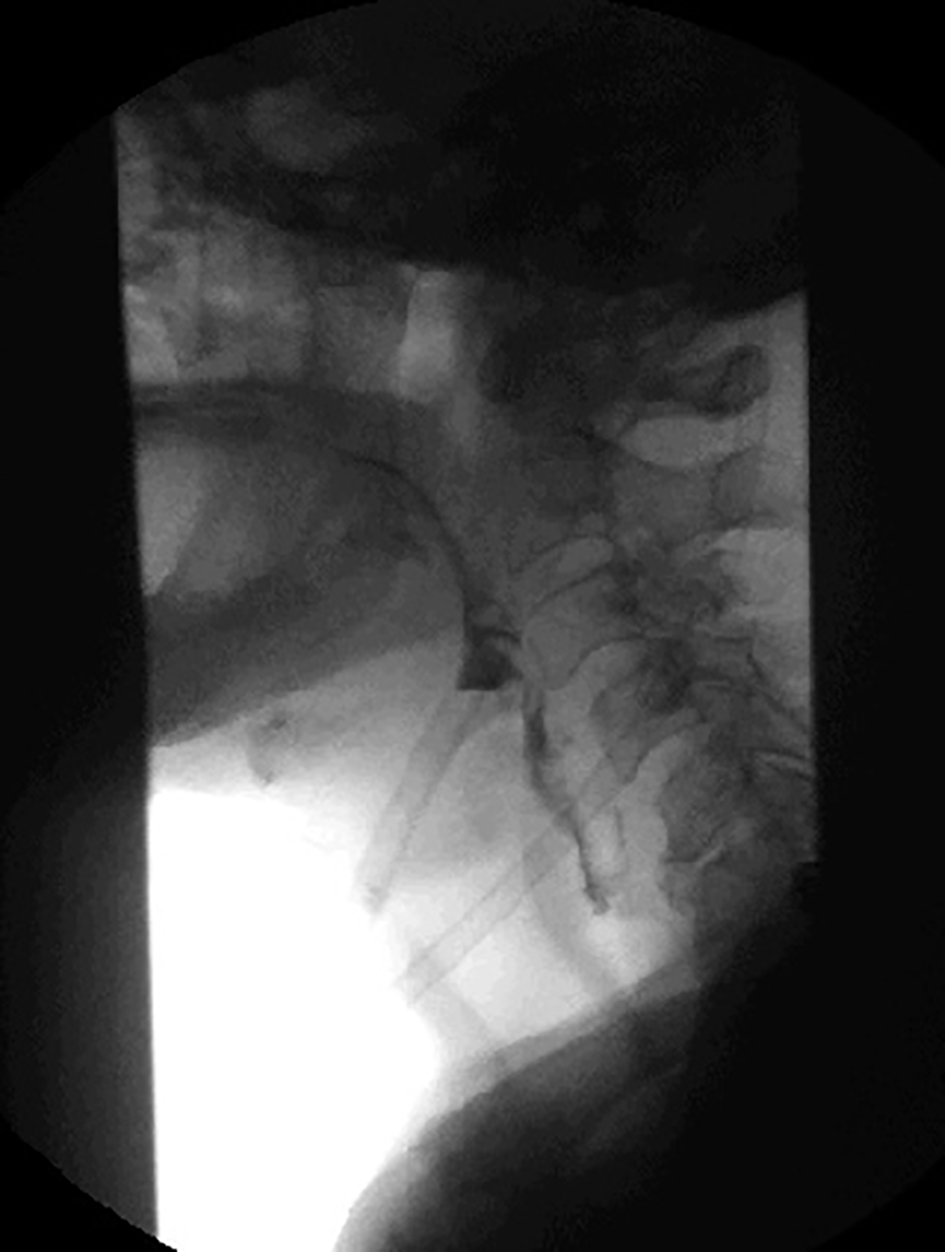 Click for large image | Figure 3. Modified barium swallow showing deep laryngeal penetration with frank tracheal aspiration. |
| Discussion | ▴Top |
OPMD is a rare genetic myopathy with adulthood onset, most often between 40 and 60 years of age. The prevalence of OPMD is highest in the French-Canadian population of the province of Quebec and among Bukhara Jews living in Israel. In the United States, the majority of affected individuals are of French-Canadian extraction, Ashkenazi Jews, and the Hispanic population [1, 2]. In Europe, the estimated prevalence is 1:100,000 [1, 3]. OPMD is a rare form of genetic muscular dystrophy which is inherited mostly as autosomal dominant and rarely as an autosomal recessive disorder. OPMD is characterized by ocular and pharyngeal muscle involvement, and it commonly presents with adulthood onset ptosis, dysphagia and proximal muscle weakness in advanced cases along with a positive family history of ptosis and dysphagia [1, 2, 4]. Our patient had all the classic clinical features of OPMD, with positive family history along with proximal muscle weakness in upper extremity which points towards advance stage of disease but was not diagnosed until late as OPMD is easily overlooked and underdiagnosed. The PABPN1 gene mutation contains GCG trinucleotide repeats, and the expansion of this repeat leads to OPMD, which was detected in our patient. The confirmatory diagnosis is made by detection of an expansion of a GCG trinucleotide repeat in the first exon of PABPN1 [5]. Muscle biopsy is warranted only in individuals with suspected OPMD who have two normal PABPN1 alleles. Unique tubulofilamentous intranuclear inclusions are seen in muscle biopsy [4, 5]. Currently, treatment is supportive as no therapeutic intervention is currently available for patients with OPMD. Surgical interventions like blepharoplasty and cricopharyngeal myotomy can temporarily help manage symptoms related to the ptosis and dysphagia, respectively. Malnutrition and recurrent aspiration pneumonia are the potential complications that reduce the life expectancy of these patients [2, 6, 7]. PABPN1 gene therapy for OPMD is in the experimental phase. The recently published article showed that the treatment of mouse model of OPMD with PABPN1 gene therapy significantly reduces the muscle fibrosis, level of insoluble aggregates, reverts muscle strength and normalizes the muscle transcriptome. The efficacy of this gene therapy was further confirmed in cells derived from OPMD patients. These findings show a promising pathway towards a gene replacement approach for OPMD treatment in future [8].
Conflict of Interest
All the participating authors disclose no conflict of interest.
Funding
None.
Authorship
All authors had access to the data and played a role in writing and critical review of this manuscript.
Consent
Yes.
| References | ▴Top |
- Abu-Baker A, Rouleau GA. Oculopharyngeal muscular dystrophy: recent advances in the understanding of the molecular pathogenic mechanisms and treatment strategies. Biochim Biophys Acta. 2007;1772(2):173-185.
doi pubmed - Brais B, Rouleau GA, Bouchard JP, Fardeau M, Tome FM. Oculopharyngeal muscular dystrophy. Semin Neurol. 1999;19(1):59-66.
doi pubmed - Fan X, Rouleau GA. Progress in understanding the pathogenesis of oculopharyngeal muscular dystrophy. Can J Neurol Sci. 2003;30(1):8-14.
doi pubmed - Brais B. Oculopharyngeal muscular dystrophy: a late-onset polyalanine disease. Cytogenet Genome Res. 2003;100(1-4):252-260.
- Brais B, Bouchard JP, Xie YG, Rochefort DL, Chretien N, Tome FM, Lafreniere RG, et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet. 1998;18(2):164-167.
doi pubmed - Codere F. Oculopharyngeal muscular dystrophy. Can J Ophthalmol. 1993;28(1):1-2.
- Coiffier L, Perie S, Laforet P, Eymard B, St Guily JL. Long-term results of cricopharyngeal myotomy in oculopharyngeal muscular dystrophy. Otolaryngol Head Neck Surg. 2006;135(2):218-222.
doi pubmed - Malerba A, Klein P, Bachtarzi H, Jarmin SA, Cordova G, Ferry A, Strings V, et al. PABPN1 gene therapy for oculopharyngeal muscular dystrophy. Nat Commun. 2017;8:14848.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.