

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 11, Number 8, August 2020, pages 253-255
A Patient With Human Herpesvirus 8-Positive Multicentric Castleman’s Disease Who Met Criteria for TAFRO Syndrome: Controversy in Practice?
Maria Cynthia Fuentes-Lacouturea, c, Carmen C. Gomeza, Martha Romerob, Andres Bordaa, Ivan Norenaa, Julian A. Naranjoa
aFundacion Cardioinfantil, Bogota D.C., Colombia
bHospital Universitario Fundacion Santa Fe de Bogota, Bogota D.C., Colombia
cCorresponding Author: Maria Cynthia Fuentes-Lacouture, Fundacion Cardioinfantil, Bogota D.C., Colombia
Manuscript submitted June 8, 2020, accepted June 16, 2020, published online July 21, 2020
Short title: TAFRO Syndrome in HHV-8-Positive MCD
doi: https://doi.org/10.14740/jmc3523
| Abstract | ▴Top |
Multicentric Castleman’s disease (MCD) is a known entity with characteristics of lymphoproliferative syndrome, characterized by cytokine activation. Its association with human immunodeficiency virus (HIV) is frequently described, as well as its positivity for human herpesvirus 8 (HHV-8). However, some negative patients for the latter (called idiopathic MCD), may have an aggressive spectrum of the disease (characterized by the presence of cytopenia, renal failure, anasarca and organomegaly), known as TAFRO syndrome (thrombocytopenia, anasarca, myelofibrosis, renal dysfunction, and organomegaly). We present the case of a young patient recently diagnosed with HIV infection, in whom MCD was found, and with an aggressive course despite treatment, who met criteria for TAFRO syndrome despite HHV-8 positivity.
Keywords: Multicentric Castleman’s disease; HHV-8; TAFRO syndrome
| Introduction | ▴Top |
Multicentric Castleman’s disease (MCD) is a common entity in patients with human immunodeficiency virus (HIV) infection. Since 2010, TAFRO syndrome that originally exists in patients with idiopathic MCD (iMCD), and characterized by thrombocytopenia, anasarca, myelofibrosis, renal dysfunction, and organomegaly [1], has been reported in many cases in literature, with main population in Japanese patients. Importantly, diagnosis of TAFRO syndrome should first exclude other entities with similar clinical manifestations as malignancies, autoimmune disorders including immune-mediated thrombocytopenia, and infectious diseases [1].
However, taking into account the multiple similarities in terms of signs and symptoms of patients suffering from TAFRO compared to other entities, some authors have suggested that this syndrome is not exclusive to iMCD, but that it could also appear in human herpesvirus 8 (HHV-8)-positive patients, as well as in those with autoimmune diseases. Furthermore, the possibility that TAFRO syndrome may be an entity other than MCD has been also raised [2].
| Case Report | ▴Top |
A 28-year-old male without past medical history was diagnosed with HIV infection after an emergency room (ER) visit for severe anemia. Two months after initial diagnosis, he was admitted to the hospital due to asthenia and persistent disabling fatigue, with occasionally fever and night sweats. At this time, highly active anti-retroviral therapy had not started yet. Physical examination was positive for fever, generalized lymphadenopathy, severe hepatosplenomegaly, and peripheral edemas. Initial studies showed severe normocytic anemia and thrombocytopenia. Lactic dehydrogenase was elevated; ferritin level was 10,443 ng/mL. Kidney and liver function test were normal upon admission. Because of peripheral edemas a transthoracic echocardiogram was performed, in which mild pericardial effusion was revealed. Neck and thoracoabdominal CT confirmed the presence of multiple lymphadenopathies and severe hepatosplenomegaly. Additional tests for Histoplasma, Cryptococcus, hepatitis B and C, Epstein-Barr virus, and cytomegalovirus were all negative. The HIV viral load at this time was 1,127,195 copies/mL, and cluster of differentiation 4 (CD4)+ lymphocyte count by flow cytometry was 144 cells/mm3. The patient was under pharmacological prophylaxis for infectious diseases. Under a suspicion of lymphoproliferative disease, cervical lymph node excisional biopsy was taken, which reported atypical lymphoid infiltrate and hyperplasia of follicular pattern with interfollicular plasmacytosis (Fig. 1a), compatible with MCD. Immunohistochemistry was positive for HHV-8 (Fig. 1b). Treatment with rituximab 375 mg/m2/weekly for 4 weeks was started. After first dose the patient developed anasarca, his hemoglobin mean level was 6 g/dL requiring daily transfusions, and thrombocytopenia worsened with a daily count < 50,000/mm3. Other laboratories revealed erythrocyte sedimentation rate (ESR) 45 mm/h, albumin 2 g/dL. Control images showed persistent hepatosplenomegaly. Despite HHV-8 positivity, TAFRO syndrome was suspected. Treatment with rituximab was continued taking into account MCD, but with a poor response and progressive deterioration, the patient died 4 months after diagnosis.
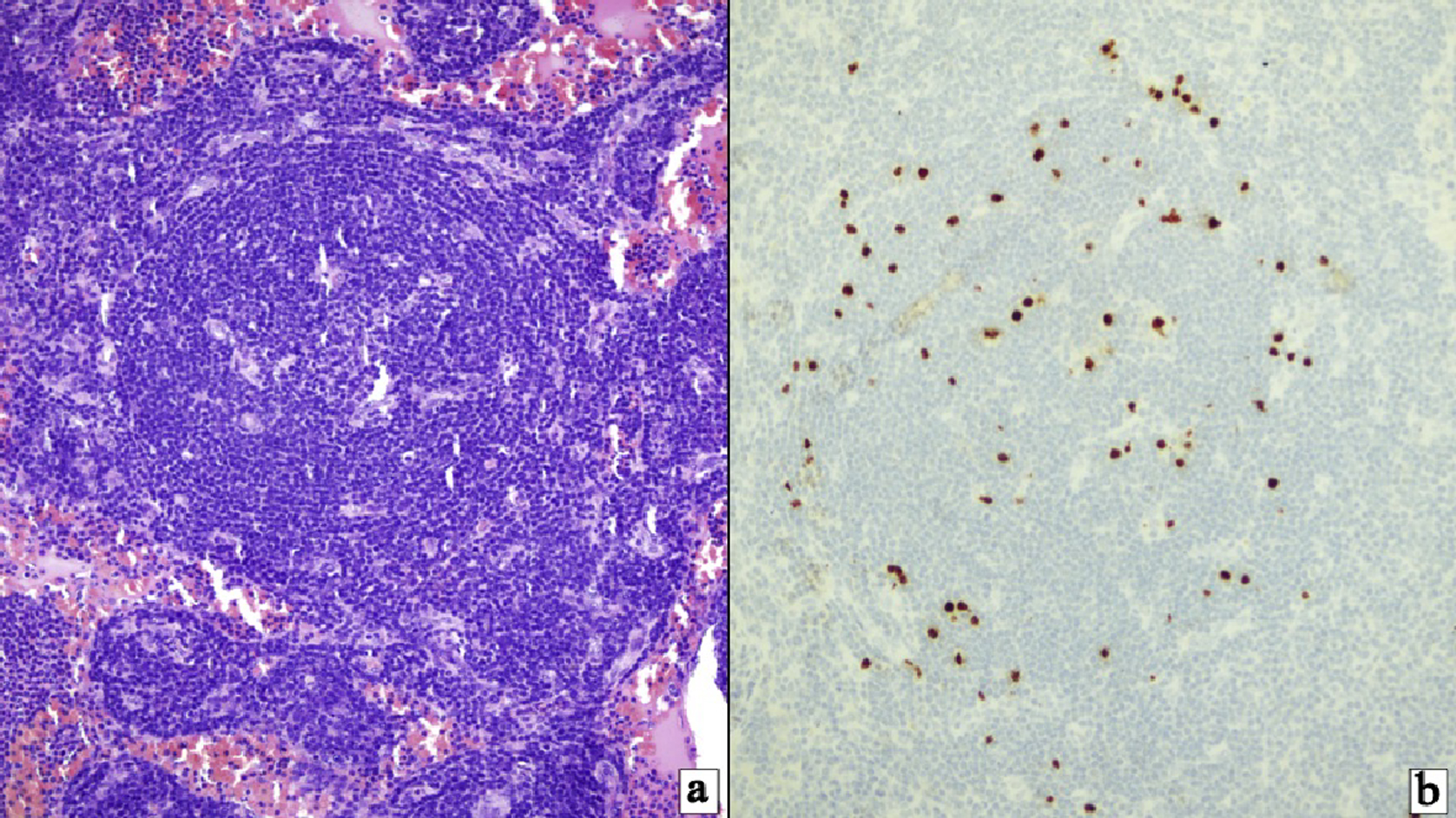 Click for large image | Figure 1. (a) HHV-8-positive multicentric Castleman’s disease. Follicles with mantle zone B cells exhibiting target-like features surrounding follicular center with radially penetrating vasculature (hematoxylin and eosin stain, × 400). (b) HHV-8-positive/KSHV infected cells identified principally within the mantle cell zones (immunostaining for HHV-8 (latent nuclear antigen-1), × 200). HHV-8: human herpesvirus 8; KSHV: Kaposi’s sarcoma-associated herpesvirus. |
| Discussion | ▴Top |
MCD is a lymphoproliferative disorder associated with HIV infection. Etiologically it has been divided into HHV-8-positive or idiopathic, taking into account the co-infection by this agent, respectively [3]. Over time, some patients with idiopathic variant presented with an aggressive spectrum of the disease, with the presence of cytopenia, anasarca, fever, bone marrow fibrosis and organomegaly. Due to this observation, in 2010 it was first described a new associated entity called TAFRO syndrome, also known as Castleman-Kojima disease, and known for having a more aggressive clinical course and early death [3].
Based on the initial descriptions, in 2015 27 members of the TAFRO research group attended a meeting where disease characteristics and new diagnostic criteria were described [3]. This model includes major and minor criteria based on the presence of anasarca, thrombocytopenia and systemic inflammation, as well as renal insufficiency, organomegaly and reticulin on bone marrow biopsy. Among all the manifestations, thrombocytopenia is the most frequent, reaching 100% of cases. Additionally, a severity score is proposed according to the commitment of each of the systems involved. In addition, therapeutic possibilities were raised, where glucocorticoids take first place, followed by other alternatives such as cyclosporin A, and biological therapy against interleukin 6 (IL-6) (tocilizumab) and CD20 (rituximab) [3].
Later in 2016 Iwaki et al published a diagnostic consensus in which patients with autoimmune diseases and co-infection with HHV-8 and Epstein-Barr virus must be first excluded before making the diagnosis of this new syndrome [4].
However, several cases have been reported in the medical literature, interrogating the possible coexistence of TAFRO syndrome in other neoplastic or autoimmune entities, sharing the pathophysiology characterized by the presence of raised cytokine levels, mediated by IL-6, vascular endothelial growth factor (VEGF), and natural killer (NK) lymphocytes [5]. Interestingly, some other research has found that blockade of IL-6 pathway does not reverse the disease as other drugs as cyclosporine A, raising the possibility of other pathophysiological mechanisms involved in this new entity, for example T cell activation in response to IL-2 [6].
Despite all the effort for describing and to understand this new disease, TAFRO syndrome remains a diagnostic challenge, with a generally aggressive course and adverse outcomes. We strongly believe there is a biologically plausible theory approving the existence of TAFRO syndrome associated with other entities in addition to iMCD, and its early recognition is useful and necessary to direct the patient to a more intensive therapy based on the aforementioned pathophysiology.
The knowledge of these cases will help us to better characterize this new disease, in order to learn to recognize it when it occurs in our practice, and finally offer future therapeutic alternatives that positively impact the evolution of the disease, thus achieving better outcomes.
Conclusions
TAFRO syndrome is a constellation of clinical and paraclinical manifestations originally described in patients with Castleman’s disease (idiopathic variant). However, cases like ours, in which the patient meets exclusion and inclusion criteria have also been described, questioning about the nature of this new entity, and making possible this new syndrome instead of being a subgroup of iMCD, may be a separate disease with multiple overlapping manifestations of other diseases. We present the case of a patient with a diagnosis of HIV infection and MCD, in whom all the criteria for TAFRO syndrome were documented, despite being co-infected with HHV-8.
Acknowledgments
None to declare.
Financial Disclosure
No funding was received.
Conflict of Interest
None to declare.
Informed Consent
The manuscript has been sufficiently de-identified to protect the patient. However, prior to death, his informed written consent was obtained for the publication of the case.
Author Contributions
All authors contributed to the editing of the manuscript. MCFL wrote the manuscript. MR made the accompanying figure.
Data Availability
The authors declare that data supporting the findings of this study are available within the article
| References | ▴Top |
- Kawabata H, Takai K, Kojima M, Nakamura N, Aoki S, Nakamura S, Kinoshita T, et al. Castleman-Kojima disease (TAFRO syndrome) : a novel systemic inflammatory disease characterized by a constellation of symptoms, namely, thrombocytopenia, ascites (anasarca), microcytic anemia, myelofibrosis, renal dysfunction, and organomegaly : a status report and summary of Fukushima (6 June, 2012) and Nagoya meetings (22 September, 2012). J Clin Exp Hematop. 2013;53(1):57-61.
doi pubmed - Carbone A, Pantanowitz L. TAFRO syndrome: An atypical variant of KSHV-negative multicentric Castleman disease. Am J Hematol. 2016;91(2):171-172.
doi pubmed - Masaki Y, Kawabata H, Takai K, Kojima M, Tsukamoto N, Ishigaki Y, Kurose N, et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Int J Hematol. 2016;103(6):686-692.
doi pubmed - Iwaki N, Fajgenbaum DC, Nabel CS, Gion Y, Kondo E, Kawano M, Masunari T, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J Hematol. 2016;91(2):220-226.
doi pubmed - Louis C, Vijgen S, Samii K, Chalandon Y, Terriou L, Launay D, Fajgenbaum DC, et al. TAFRO syndrome in Caucasians: a case report and review of the literature. Front Med (Lausanne). 2017;4:149.
doi pubmed - Sakashita K, Murata K, Takamori M. TAFRO syndrome: current perspectives. J Blood Med. 2018;9:15-23.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.