

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 12, Number 5, May 2021, pages 195-201
Pancreatic VIPoma as a Differential Diagnosis in Chronic Pediatric Diarrhea: A Case Report and Review of the Literature
Carolina Bonilla Gonzaleza, d, Juliana Rusinqueb, Camila Uribeb, Ailim Cariasc, Maria Luisa Contrerasa
aPediatric Critical Care, University Hospital Fundacion Santa Fe de Bogota, Bogota, Columbia
bUniversidad de los Andes, Bogota, Colombia
cPediatric Gastroenterology, University Hospital Fundacion Santa Fe de Bogota, Bogota, Columbia
dCorresponding Author: Carolina Bonilla Gonzalez, University Hospital Fundacion Santa Fe de Bogota, Av. 9 No 116-20 Consultorio 617, Bogota 110111, Colombia
Manuscript submitted November 8, 2020, accepted November 26, 2020, published online March 5, 2021
Short title: VIPoma as a Diagnosis in Pediatric Diarrhea
doi: https://doi.org/10.14740/jmc3535
| Abstract | ▴Top |
Chronic diarrhea is a common chief complaint in the pediatric population with a wide range of diagnostic differentials; as such, whilst suspecting common causes, less prevalent conditions tend to be overlooked, such as neuroendocrine tumor pathologies. VIPomas are characterized by hypersecretion of the vasoactive intestinal peptide (VIP), causing watery diarrhea, hypokalemia, and achlorhydria. Nonetheless, its low incidence rate in children makes it an easily unnoticed pathology. Herein, we report a case of a 14-year-old female patient and a review of relevant literature. The patient complained of 7-month history of watery diarrhea, multiple emetic episodes, and relevant past medical history of multiple hospitalizations. Chronic diarrheal disease work-up studies, including a high VIP scintigram, showed a lesion suggestive of a VIPoma-type neuroendocrine pancreatic tumor. A distal pancreatectomy was performed with a complete resolution of the symptoms. When faced with a pediatric patient presenting with chronic secretory diarrhea and whose work-up studies rule out the most common pathologies, the possible presence of a neuroendocrine tumor as VIPoma should be considered.
Keywords: Chronic diarrhea; Vasoactive intestinal peptide; Pancreatic tumor; Pediatric
| Introduction | ▴Top |
The incidence of neuroendocrine tumors (NETs) is low, with only 2.8 cases per a million inhabitants reported in patients under 30 years of age [1]. Moreover, the reports of accurate diagnoses in the pediatric population are even more sporadic [2]. NETs secrete vasoactive substances such as peptides and neurotransmitters in various tissues, including the lungs, bronchial tree, thymus, gonads, thyroid and/or gastrointestinal tract [3]. These tumors are classified as functional and nonfunctional. The former, depending on their location, induce hypersecretion of specific hormones leading to characteristic symptoms [3]. In patients under 20 years old, NETs of functioning pancreatic localization (PNET) represent 30% of all NETs [4], with the most frequent causes representing the insulinoma, gastrinoma, glucagonoma, VIPoma and somatostatinoma [4, 5] (Fig. 1).
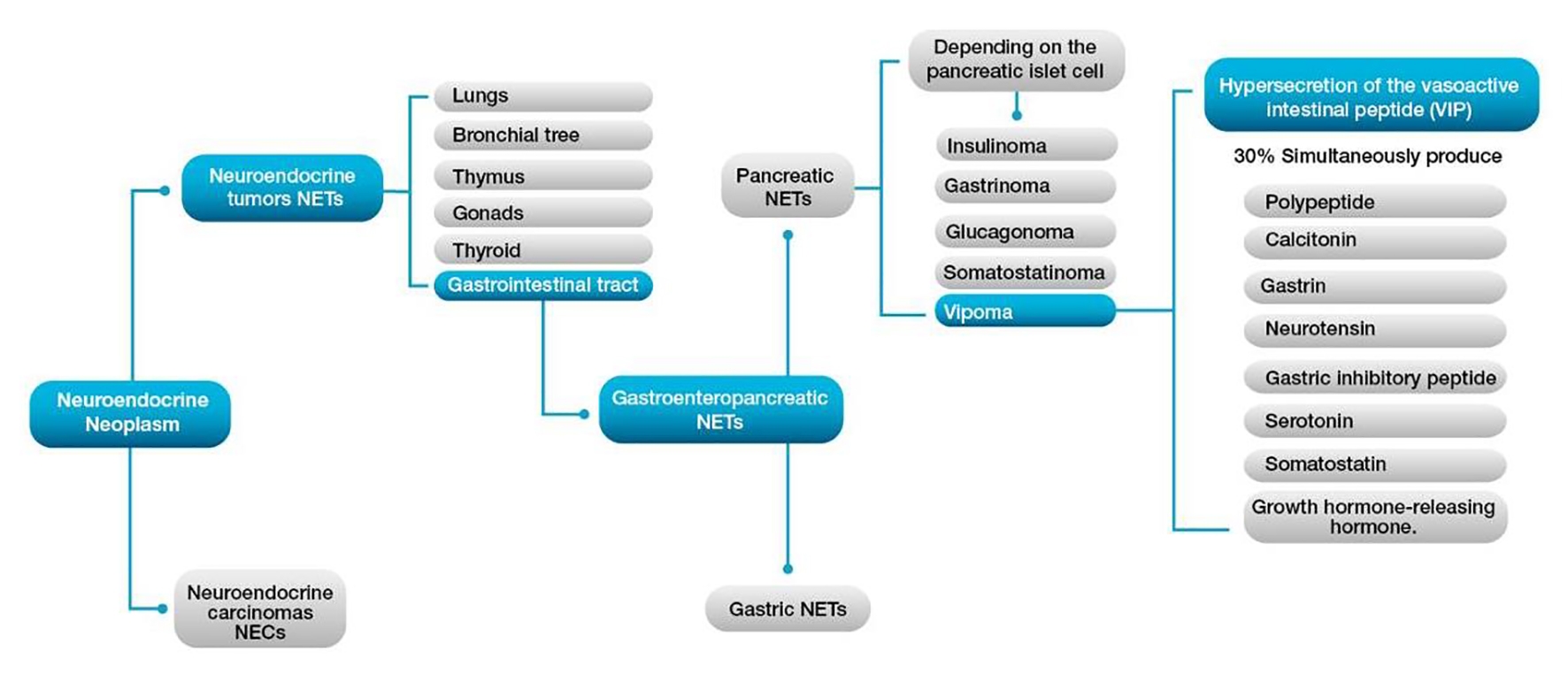 Click for large image | Figure 1. Clinical classification of neuroendocrine tumors (NETs). |
Pancreatic VIPomas represent 3-8% of all pancreatic NETs, with an incidence of 0.2 - 0.5 case/1,000,000 inhabitants [6]. As suggested by their names, these tumors are vasoactive intestinal peptide (VIP) secretors, and up to 30% of them simultaneously produce pancreatic polypeptide, calcitonin, gastrin, neurotensin, gastric inhibitory peptide, serotonin, somatostatin, and a growth hormone-releasing hormone (Fig. 2). The VIPoma can present itself as an isolated neoplasm or, exceptionally, be associated with multiple endocrine neoplasia types 1 (MEN1) [6].
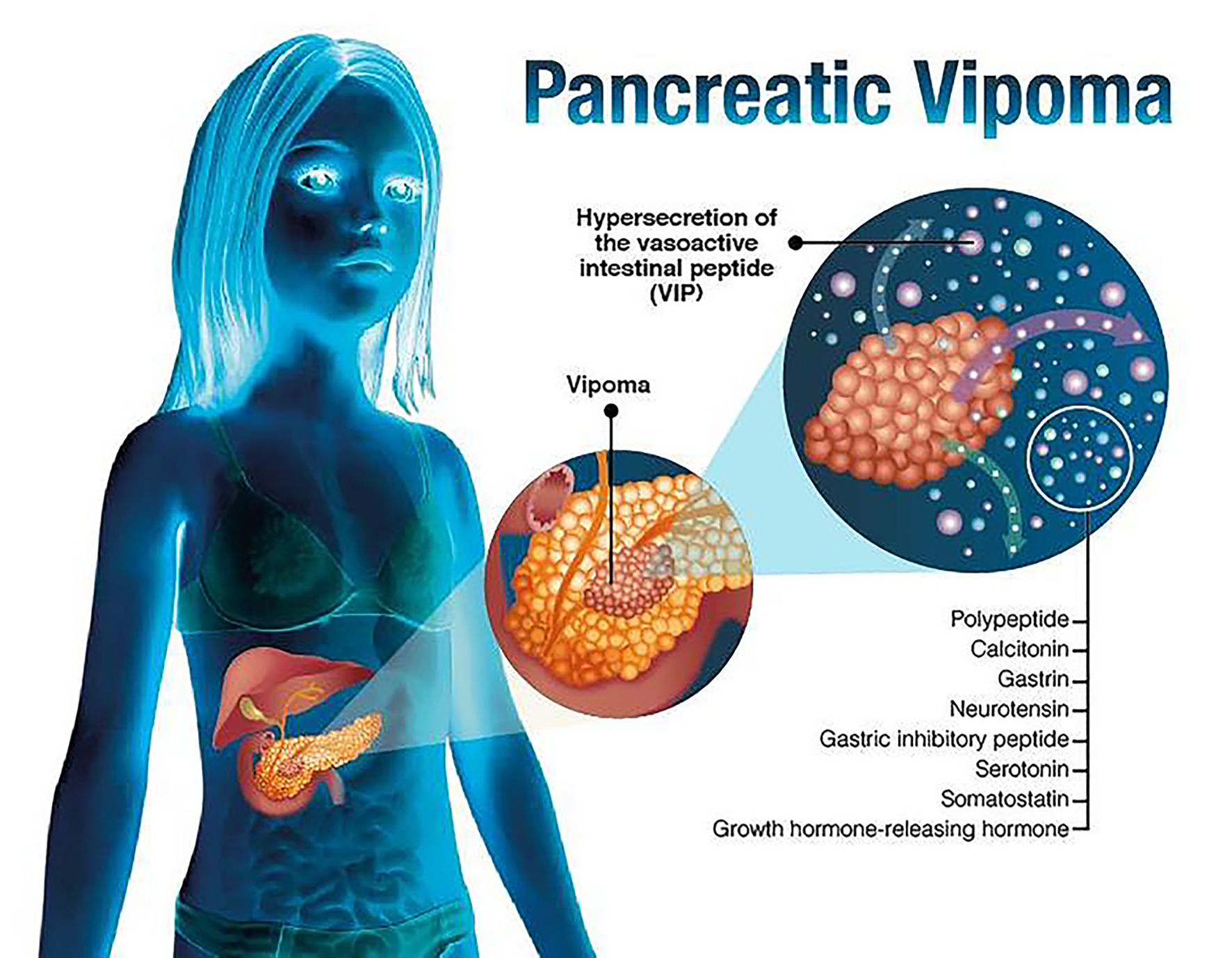 Click for large image | Figure 2. Pancreatic VIPoma. |
Generally, the VIPoma is described as watery diarrhea, hypokalemia, and achlorhydria (WDHA) syndrome (or “pancreatic cholera”), characterized by watery diarrhea (> 7 L/day), hypokalemia and achlorhydria. Pancreatic tumors are usually reported as a unique and symptomatic mass, greater than 3 cm in size, located in the tail of the pancreas in 75% of cases and with high incidence of metastasis at the time of diagnosis (up to 60-80%), particularly to lymph nodes, liver, kidneys or bone [7].
| Case Report | ▴Top |
A 14-year-old female patient with no prior medical history presented to the hospital complaining of 7 months of watery diarrhea, approximately 7 stools/day, associated with poorly characterized abdominal pain, vomiting, hypoxia, arthralgia, myalgia and unintentional weight loss of 11 kg. Initial work-up studies revealed hypokalemia (1.6 mEq/L) associated with electrocardiographic changes, requiring initial intravenous potassium chloride administration. Stool cultures and serologic testing for common bowel diseases were negative, and immune profile testing was completely normal: immunoglobulin A (IgA) 1.23 U/mL, negative antinuclear antibodies (ANAs), negative antineutrophil cytoplasmic antibodies (ANCAs), gliadin antibodies immunoglobulin G (IgG) 2.67 U/mL, IgG antibodies transglutaminase 23 U/mL, and IgA gliadin 3.42 U/mL. Similarly, bone scan, endoscopy and colonoscopy all revealed normal results.
Endocrinological profile was evaluated, with the following results: free T4 0.76 ng/dL, cortisol am 13.71 µg/dL, pm 5.02 µg/dL, adrenocorticotropic hormone (ACTH) 10.8 pg/mL, aldosterone 65 pg/mL, and thyroid stimulating hormone (TSH) 1.73 µIU/mL (all in normal ranges). Additionally, the patient had normal gastrin levels (17.9 pg/mL), with a low parathyroid hormone at 4.5 pg/mL, and serum copper 148 µg/dL (normal), zinc 84 µg/dL (normal), alpha 1 antitrypsin in stool 2 mg/g (normal), with an important VIP elevation: value of 91.2 pmol/L.
Due to the identified VIP elevation, a scintigraphy was performed, demonstrating a pancreatic lesion with prominent octreotide uptake. The patient was hospitalized in the pediatric intensive care unit (PICU) due to persisting high fecal expenditure of about 7 to 10 L/day and severe hypokalemia, with important dehydration and electrolyte inbalance. Magnetic resonance imaging (MRI) scans were performed, which identified two high uptake focal lesions adjacent and above the posterior aspect of the pancreas body, measuring 20 ×16 mm and 10 × 9 mm, with enhancement similar to that of pancreatic tissue, which correlates with focal uptake scintigraphy of octreotide compatible with a clinical diagnosis of VIPoma (Fig. 3).
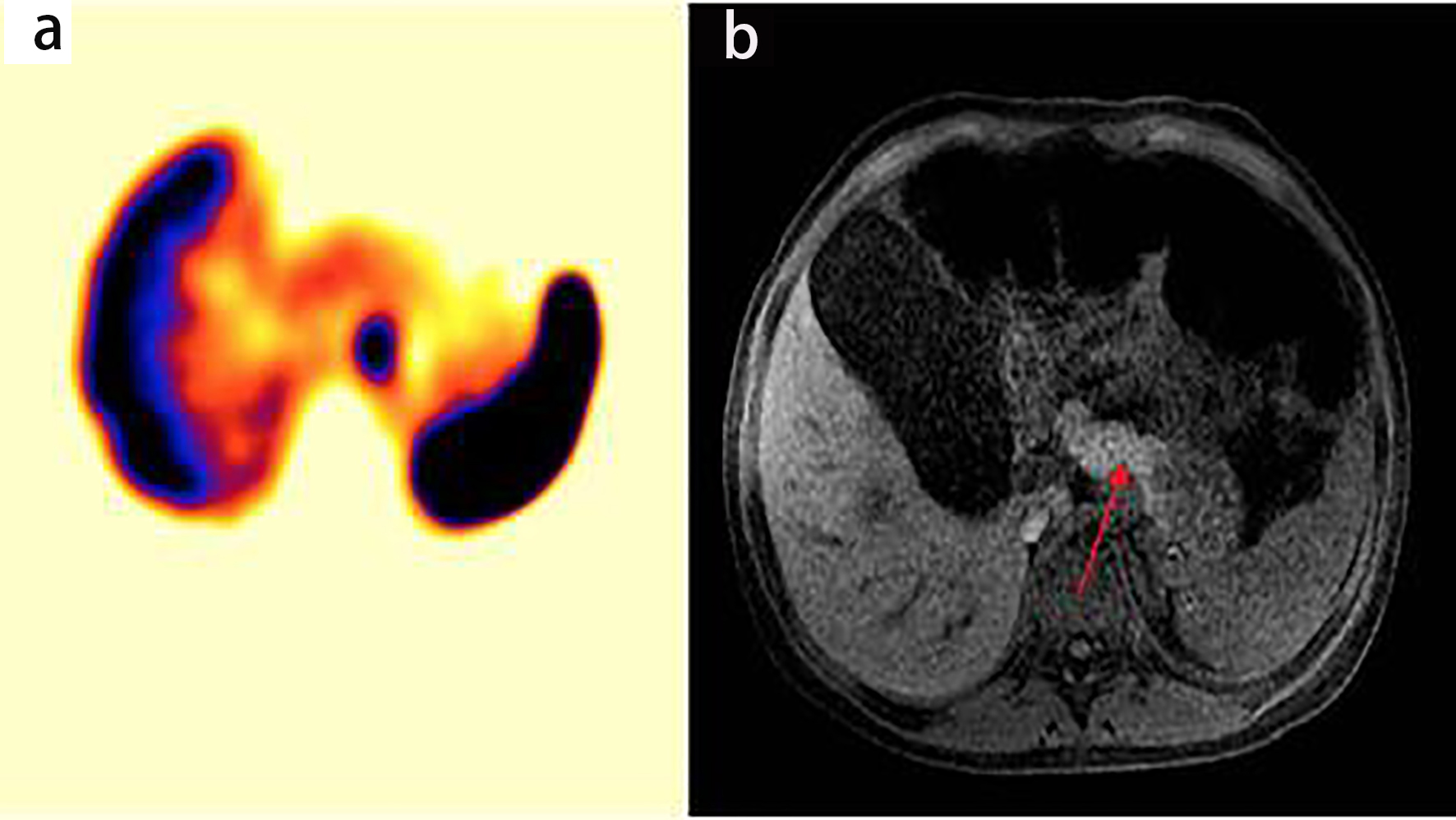 Click for large image | Figure 3. (a) Octreotide scintigraphy showing hyperintense lesion pancreas. (b) Magnetic simple and proven abdomen showing focal lesions solid appearance adjacent to the posterior and superior aspect of the pancreas body (arrow). |
The biliopancreatic endoscopic ultrasound reported a normal pancreatic head echo-structure without observable injury. In the pancreatic body, four hypoechoic lesions were observed. These were described as homogeneous, round, well defined, with the largest measuring 13 mm and the smallest 7 mm in diameter. All reported lesions were compatible with NETs. A fine-needle aspiration biopsy was performed by transgastric route, obtaining samples of the largest lesion. Immunohistochemistry studies confirmed positivity for CAM5.2, synaptophysin, and chromogranin, confirming the diagnosis.
Initial management with octreotide resulted in dramatic clinical improvement. Subsequent distal pancreatectomy was performed, confirming the four tumor lesions described previously in the proximal body of the pancreas. The resected pancreatic tissue was 9.5 × 5 × 2.9 cm, of which 0.5 × 0.3 cm was immunohistochemically well differentiated (which primarily involved the proximal body and tail of the pancreas). Metastatic involvement was present in four of the nine resected peri-pancreatic lymph nodes.
Microscopically, the tumor was characterized by cellular proliferation with slight nuclear pleomorphism and scant cytoplasm, arranged in small trabeculae and pseudoacinar formations with perineural invasion, without endangering the section edge. Mitotic rate was below 2 mitoses/mm2.
Immunohistochemistry showed a reaction for synaptophysin, chromogranin and cytokeratin 19. Expression was not observed for insulin, glucagon, somatostatin, serotonin, gastrin or companion diagnostics. The reported Ki-67 index was 6.9%.
Using the World Health Organization (WHO) Stage III and the American Joint Committee on Cancer (AJCC) metastasis classification systems, a final diagnosis of a functional pancreatic primary NET VIPoma Grade 2 was made (based on the tumor’s clinical, laboratory, radiologic, and histopathologic (immunohistochemical) profile). Further classification of VIPomas is detailed in Table 1, using the latest WHO 2017 classification system, in which final grade is determined based on whichever index (Ki-67 or mitotic) places the tumor in the highest-grade category [8]. After surgery, the patient presented symptomatic improvement, with complete resolution of diarrhea, and correction of electrolyte/acid-base profiles. A post-operative MRI showed a residual lesion in the sella turcica, consisting of an incidental focal lesion in the pituitary gland, later classified as a small self-limited bleeding Rathke pouch cyst, which will be followed up in an outpatient setting. The patient was subsequently discharged after 10 days of in-hospital management.
 Click to view | Table 1. VIPoma Classification |
| Discussion | ▴Top |
To develop an adequate review of the literature, a manual search was conducted in Medline and PubMed of cases reported in children younger than 18 years old using the Medical Subject Headings (MeSH) terms: “VIPoma”, “pancreas”, “diarrhea”, “neuroendocrine tumors”, “children”, and “chronic diarrhea”. Out of a total of 60 articles, 23 were reviewed as compliant with search criteria, including five case reports to be analyzed.
In reviewing published literature, we found twenty-three reported cases of pediatric patients presenting with chronic diarrhea and elevated serum VIP. Seven (30.4%) of these cases were ganglioneuroblastomas, eight (34.8%) ganglioneuromas, three (13.0%) were not histopathologically identified, and five (21.7%) included pancreatic VIPomas. The latter cases of children diagnosed with pancreatic VIPoma were analyzed. The reported female to male ratio was 3:2, with the earliest age at onset at seven months and the oldest patient presenting symptoms at fifteen years of age. The predominant symptom in all four cases was diarrhea with high expenditure, from 2 to 10 L/day, with no abnormalities present in the physical examination. Other associated symptoms were vomiting, redness, dehydration, pallor and light headedness.
The treatment for the five reported cases was surgical resection, involving distal pancreatectomy, and two cases included a splenectomy in the same surgical procedure. Metastases were identified in three cases (60%), distributed in the adjacent lymph nodes, liver and spleen.
All the cases reported an improvement in diarrhea after surgery and normal subsequent VIP and potassium levels, with a resolution of acidosis. The 7-month-old patient [9] presented a gastric fistula and post-operative sepsis development, ultimately leading to death. The 11-year-old patient [10] had a normal post-operative period, completely asymptomatic, and with magnetic resonance showing no abnormality at 18 months of follow-up. The 12-year-old patient [11] required adjunctive octreotide management 3 months following surgery, with a worsening of symptoms 5 months after surgery which required the addition of sunitinib. After 13 months of initiation of therapy, doctors found a metastatic mediastinal VIPoma, which required replacing medical management to chemotherapy (temozolomide). The 15-year-old patient [12] presented gastrointestinal bleeding that required blood, yet the symptoms ceased, and the patient was discharged at the nineteenth day of hospitalization and presented symptom-free at her 36-month follow-up appointment. Finally, in the last case report of Rastogi et al [13], a 2-year-old patient was started on intravenous octreotide therapy, showing important significant symptomatic improvement. She was then subject to laparotomy for VIPoma resection, with intra-operative potassium replacement and central venous pressure-guided fluid therapy. The patient was discharged symptom-free on the 10th post-operative day. Further details and comparisons in patient characteristics and clinical course are detailed in Table 2 [9-13].
Click to view | Table 2. VIPoma Clinical Characteristics in Pediatric Patients Across Published Case Reports |
Pancreatic neuroendocrine tumors (panNETs) are classified clinically as nonfunctional or functional, based on the properties of the hormones they secrete and their ability to produce a clinical syndrome. Moreover, functional panNETs are much less common and present with specific clinical syndromes depending on the secreted hormone. In this case, VIP is responsible for regulating intestinal motility, stimulating adenylate cyclase and driving the secretion of electrolytes and water from the gastrointestinal mucosa [14]. The WDHA syndrome (“pancreatic cholera”), first described by Verner and Morrison in 1958, is characterized by chronic watery diarrhea, hypokalemia, and hyperchlorhydria or achlorhydria in association with pancreatic tumors [15], being the main clinical picture present in most VIPoma cases.
In the pediatric population, VIPomas most frequently arise from the bronchus, colon, liver, and neural crest-derived tissues (e.g. sympathetic nerve chain, pituitary, thyroid, and adrenal glands), and rarely originate from the pancreas [16, 17].
PanNETs are more prevalent in adults, and as such, reported pediatric cases are scarce [18]. This present pediatric case is, to our knowledge, the sixth published case worldwide and the first reported in Colombia. All disclosed cases thus far have presented the typical clinical manifestations: persistent, high-volume diarrhea that does not improve with oral intake or medications, along with hypokalemia and achlorhydria. Diarrhea is characterized by exceeding 3 L/day, odorless, free of blood, mucus, with high sodium and low gap osmolality [19].
VIPoma hypokalemia can be attributed to chronic, abundant diarrheal losses, the increased production of aldosterone and direct potassium secretion by enterocytes. This, in turn, is associated with other symptoms, such as muscle weakness, flaccid paralysis, abdominal cramps, respiratory depression and electrocardiographic changes. In our review, only one pediatric case reported electrocardiographic changes. Moreover, increased fecal excretion of bicarbonate occurs, leading to a hypokalemic metabolic acidosis without anion gap [20].
The inhibitory effect of VIP on parietal gastric mucosal cells leads to a secondary decrease in gastric acid production, heightening the aforementioned hyperchlorhydria, which potentiates malabsorption-associated dietary deficiencies of magnesium, zinc, and vitamins C, K, and B [21].
Other accompanying symptoms have similarly been described, including vomiting, abdominal pain, non-specific rash, facial redness and swelling, indigestion, back pain, lethargy and unintentional weight loss [19]. The final symptom was evident in our patient, who presented an unintentional 11 kg weight loss by the time we reached a diagnosis.
Both hyperglycemia and hypercalcemia may be simultaneously present in VIPoma cases, with poorly understood mechanisms. Of note, in cases where hypercalcemia arises in the context of a VIPoma associated concurrently with MEN1, hyperparathyroidism must be ruled out.
Electrolyte imbalances must be corrected upon hospital admission in order to avoid renal failure secondary to dehydration. Approximately 60-80% of cases have metastasis at the time of diagnosis, with liver, lymph nodes, lungs and kidneys being the most popular tissues compromised [22].
A strong clinical suspicion is necessary in order to request specific metabolic tests, such as VIP, gastrin, and glucagon (along with an electrolyte panel including potassium, sodium, and chlorine) [16, 17]. Most reported cases present with VIP serum levels between 60 and 2,100 pg/mL, yet values higher than 200 pg/mL are diagnostic.
Both anatomic and molecular imaging are important for clarifying tumor location and characteristics [12]. Neoplastic masses that surpass 1 cm in diameter can be identified directly by simple computed tomography (CT) (with a sensitivity between 85% and 100%) or with MRI (with a reported sensitivity of 75% to 100%) [4, 5]. Since between 80% and 90% of all VIPomas are positive receptors for somatostatin, we used scintigraphy to diagnose our patient accurately. This molecular imaging method has a high attraction to somatostatin receptors (octreotides) and a high reported sensitivity (between 78-100%). Additionally, the use of intraoperative ultrasound has a sensitivity of 83-100% in the detection of neoplasms that are not visible with the preoperative conventional imaging [23, 24].
Although immunohistochemically the VIPoma can secrete more than a single hormone, there is always a predominant hormone responsible for the clinical picture. VIPoma tumor cells stain positive for VIP (the most accurate marker), chromogranin A, synaptophysin, somatostatin, neuron-specific enolase and cytokeratin [20, 21]. These tumors are most commonly located in the tail of the pancreas, followed in high-frequency appearance in the pancreatic body. Lesions are usually greater than 3 cm in diameter at the time of diagnosis. Evidence demonstrating the benefits of surgical intervention in the pediatric population is scarce; as such, clinicians must rely on adult case reports that indicate that the gold-standard of VIPoma management, with or without metastases, is surgical resection of the lesion (with or without pancreatectomy).
Symptomatic management, both pharmacological and therapeutic, is indicated in individuals who refuse to undergo surgery or have unresectable metastases at the time of presentation. Clinical improvement can be established with somatostatin analogs (e.g. octreotide), controlling diarrhea by decreasing VIP secretion and abating VIPoma through antiproliferative properties [25].
Chemotherapy may be recommended as a first-line treatment option in certain patients with unresectable metastases at the time of presentation, either the preferred doxorubicin-streptozocin combination or 5-fluorouracil-streptozocin combination, in cases where specific doxorubicin contraindications arise. To date, there are various reports submitting poor evidence of success with either modality [26].
Subsequent follow-up after treatment (measuring serum levels of VIP and imaging) is currently recommended at the 1-year mark, and afterwards up to 10 years from the time of diagnoses [20].
Overall survival time is 96 months; however, this outcome depends on the tumor grade upon diagnosis, neoplastic staging and adequate surgical resection. Previous studies have found a survival rate of 5 years in patients with surgical resection with and without metastases between 15.5% and 61%, respectively, with still lower survival time in those presenting with in stage IV neoplasm [2, 3].
Conclusions
When faced with a pediatric patient presenting with chronic secretory diarrhea and whose work-up studies rule out the most common pathologies, the possible presence of a NET as VIPoma should be considered. As its most common clinical manifestations are aqueous diarrhea, hypokalemia and achlorhydria, initial studies should evaluate electrolyte inbalance, hormone levels and vasoactive substances associated with exogenous NET production. Moreover, diagnostic imaging tools such as simple CT and MRI are helpful aids to confirm the diagnosis and assess treatment. Surgical resection is currently considered as the treatment modality of choice, providing better prognosis.
Acknowledgments
None to declare.
Financial Disclosure
There was no commercial or financial incentive associated with the writing and publishing of the manuscript.
Conflict of Interest
None of the authors have competing interest or any conflict of interest to declare.
Informed Consent
Written informed consent was obtained from the parent (legal guardian) for publication of this case report and any accompanying images.
Author Contributions
All five authors worked collaboratively in the various stages of the writing process, including: gathering relevant patient’s information and obtaining informed consent, submitting the case to be reviewed and approved by the Institutional Ethics Committee at Hospital Universitario Fundacion Santa Fe de Bogota, writing the manuscript, developing original figures, subsequent editing and submission process.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
WDHA: watery diarrhea, hypokalemia, and achlorhydria (WDHA) syndrome (or “pancreatic cholera”); ANCAS: antineutrophil cytoplasmic antibodies; ACS: antinuclear antibodies; VIP: vasoactive intestinal peptide; NETs: neuroendocrine tumors; PNET: pancreatic neuroendocrine tumor; MEN1: multiple endocrine neoplasia type 1; PICU: pediatric intensive care service; MRI: magnetic resonance imaging
| References | ▴Top |
- Howell DL, O'Dorisio MS. Management of neuroendocrine tumors in children, adolescents, and young adults. J Pediatr Hematol Oncol. 2012;34(Suppl 2):S64-68.
doi pubmed - Diets IJ, Nagtegaal ID, Loeffen J, de Blaauw I, Waanders E, Hoogerbrugge N, Jongmans MC. Childhood neuroendocrine tumours: a descriptive study revealing clues for genetic predisposition. Br J Cancer. 2017;116(2):163-168.
doi pubmed - Massironi S, Sciola V, Peracchi M, Ciafardini C, Spampatti MP, Conte D. Neuroendocrine tumors of the gastro-entero-pancreatic system. World J Gastroenterol. 2008;14(35):5377-5384.
doi pubmed - Khanna G, O'Dorisio SM, Menda Y, Kirby P, Kao S, Sato Y. Gastroenteropancreatic neuroendocrine tumors in children and young adults. Pediatr Radiol. 2008;38(3):251-259, quiz 358-259.
doi pubmed - Chung EM, Travis MD, Conran RM. Pancreatic tumors in children: radiologic-pathologic correlation. Radiographics. 2006;26(4):1211-1238.
doi pubmed - Nikou GC, Toubanakis C, Nikolaou P, Giannatou E, Safioleas M, Mallas E, Polyzos A. VIPomas: an update in diagnosis and management in a series of 11 patients. Hepatogastroenterology. 2005;52(64):1259-1265.
- Rose S. Pancreatic neuroendocrine neoplasms F. Sessa. 2015.
- WHO classification of tumours of endocrine organs, 4th ed. Lloyd RV, Osamura RY, Kloppel G, Rosai J (Eds), IARC Press, Lyon. 2017. p. 211. Copyright 2017 International Agency for Research on Cancer.
- Ghishan FK, Soper RT, Nassif ED, Younoszai MK. Chronic diarrhea of infancy: nonbeta islet cell hyperplasia. Pediatrics. 1979;64(1):46-49.
- Samal SC, Paul AC, Venkateswari S, Nair S, Venkatramani S, Perakath B, Simon A, et al. VIPoma of pancreas in a child. Indian J Gastroenterol. 2000;19(4):194-195.
- Bourcier ME, Vinik AI. Sunitinib for the treatment of metastatic paraganglioma and vasoactive intestinal polypeptide-producing tumor (VIPoma). Pancreas. 2013;42(2):348-352.
doi pubmed - Brenner RW, Sank LI, Kerner MB, Schrager GO, Elguezabal A, Roth J. Resection of a vipoma of the pancreas in a 15-year-old girl. J Pediatr Surg. 1986;21(11):983-985.
doi - Rastogi A, Priya V, Haldar R, Riaz R. Paediatric VIPoma: A Jamboree of Electrolytes. Turk J Anaesthesiol Reanim. 2018;46(2):158-160.
doi pubmed - Anderson CW, Bennett JJ. Clinical presentation and diagnosis of pancreatic neuroendocrine tumors. Surg Oncol Clin N Am. 2016;25(2):363-374.
doi pubmed - Belei OA, Heredea ER, Boeriu E, Marcovici TM, Cerbu S, Marginean O, Iacob ER, et al. Verner-Morrison syndrome. Literature review. Rom J Morphol Embryol. 2017;58(2):371-376.
- Natanzi N, Amini M, Yamini D, Nielsen S, Ram R. Vasoactive intestinal peptide tumor. Scholarly Research Exchange. 2009;2009:1-7.
doi - Vinik A. Vasoactive Intestinal Peptide Tumor (VIPoma). In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, et al., eds. Endotext. South Dartmouth (MA), 2000.
- Ayub A, Zafar M, Abdulkareem A, Ali MA, Lingawi T, Harbi A. Primary hepatic vipoma. Am J Gastroenterol. 1993;88(6):958-961.
- Smith SL, Branton SA, Avino AJ, Martin JK, Klingler PJ, Thompson GB, Grant CS, et al. Vasoactive intestinal polypeptide secreting islet cell tumors: a 15-year experience and review of the literature. Surgery. 1998;124(6):1050-1055.
doi pubmed - Song S, Shi R, Li B, Liu Y. Diagnosis and treatment of pancreatic vasoactive intestinal peptide endocrine tumors. Pancreas. 2009;38(7):811-814.
doi pubmed - Berney C, Kurt AM, Kapanci Y. [Endocrine tumors of the pancreas: immunohistochemical and clinical correlation]. Schweiz Med Wochenschr. 1994;124(5):179-185.
- Ahmad N. Zarzour prolonged survival in metastatic to vasoactive intestinal patient with peptide producing pancreatic neuroendocrine tumors. Journal of Clinical Case Reports. 2012;2(15).
doi - Rosch T, Lightdale CJ, Botet JF, Boyce GA, Sivak MV, Jr., Yasuda K, Heyder N, et al. Localization of pancreatic endocrine tumors by endoscopic ultrasonography. N Engl J Med. 1992;326(26):1721-1726.
doi pubmed - King C, Reznek R, Dacie J, Wass J. Imaging islet cell tumors. 1994.
doi - Kraenzlin ME, Ch'ng JL, Wood SM, Carr DH, Bloom SR. Long-term treatment of a VIPoma with somatostatin analogue resulting in remission of symptoms and possible shrinkage of metastases. Gastroenterology. 1985;88(1 Pt 1):185-187.
doi - Ramage JK, Ahmed A, Ardill J, Bax N, Breen DJ, Caplin ME, Corrie P, et al. Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumours (NETs). Gut. 2012;61(1):6-32.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.