

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 11, Number 10, October 2020, pages 320-323
Acute Plasma Cell Leukemia Presenting as Primary Hyperammonemic Encephalopathy
Dveet Patela, b, d, Andrew Manganob, David Mocciab, Shawn Espertib, Kunjan Udanib, Jordan Hendersonb, Nino Balanchivadzec
aDepartment of Internal Medicine, Grand Strand Health, Myrtle Beach, SC, USA
bAffiliated Hospital of Grand Strand Medical Center, Myrtle Beach, SC, USA
cDepartment of Hematology/Oncology, Henry Ford Health System, Detroit, MI, USA
dCorresponding Author: Dveet Patel, Affiliated Hospital of Grand Strand Medical Center, 809 82nd Parkway Myrtle Beach, SC 29572, USA
Manuscript submitted August 1, 2020, accepted August 8, 2020, published online August 28, 2020
Short title: Unusual Presentation of Acute PCL
doi: https://doi.org/10.14740/jmc3559
| Abstract | ▴Top |
Primary plasma cell leukemia (PPCL) is a rare form of multiple myeloma (MM) and is a rare aggressive disease with a median overall survival of 6 - 11 months. We present a case of acute hyperammonemic encephalopathy as the initial presentation of PPCL in a 78-year-old woman to highlight an atypical presentation of this disorder.
Keywords: Plasma cell leukemia; Hyperammonemic encephalopathy; Multiple myeloma
| Introduction | ▴Top |
Primary plasma cell leukemia (PPCL) is a form of leukemia with a reported incidence of one case per million annually in the USA [1]. It is clinically and genetically distinct from multiple myeloma (MM) with a poor prognosis and dismal survival despite recent advances in treatment modalities. We present a case of acute hyperammonemic encephalopathy as the initial presentation of PPCL in an elderly woman to highlight an atypical presentation of this uncommon disorder in hopes of helping guide early diagnosis of this fatal disease.
| Case Report | ▴Top |
A 78-year-old African American woman with a past medical history of hypertension, gastroesophageal reflux disease, and iron deficiency anemia presented with shortness of breath, hypoxia, and acute encephalopathy. Two months prior to hospital admission she was found to have new thrombocytopenia (platelet count of 93 × 103/uL), in addition to normocytic anemia (hemoglobin 9.8 g/dL), which was unchanged from her baseline (9 - 10 g/dL). She was referred to a hematologist for outpatient workup, but hospital admission preceded this evaluation. On presentation to the hospital her vital signs were: temperature 37 °C, pulse 98 beats per minute, blood pressure 130/60 mm Hg, respiratory rate 40 breaths per minute, and oxygen saturation 90% on room air. Physical exam revealed a disoriented and somnolent patient with diminished reflexes in her lower extremities. Breath sounds were absent in the infrascapular area of the left chest wall. No cervical, axillary, or inguinal lymphadenopathy was appreciated. The remaining examination was unremarkable.
Laboratory investigations revealed a total leukocyte count of 19,800 × 103/µL (67% lymphocytes, 9% monocytes, 1% myelocytes, 20% segmented neutrophils), hemoglobin level of 7.8 g/dL with mean corpuscular volume 89.5, and platelet count of 32,000 × 103/µL. Comprehensive metabolic panel was unremarkable with normal renal function, electrolytes, calcium, and liver function testing. Peripheral smear demonstrated atypical lymphocytes and 38% circulating plasma cells. Ammonia level was elevated at 78 µmol/L (normal < 35 µmol/L). Coagulation studies were within normal limits. Lactate dehydrogenase (LDH) was 650 U/L. Peripheral smear revealed atypical plasma cells as seen in Figure 1.
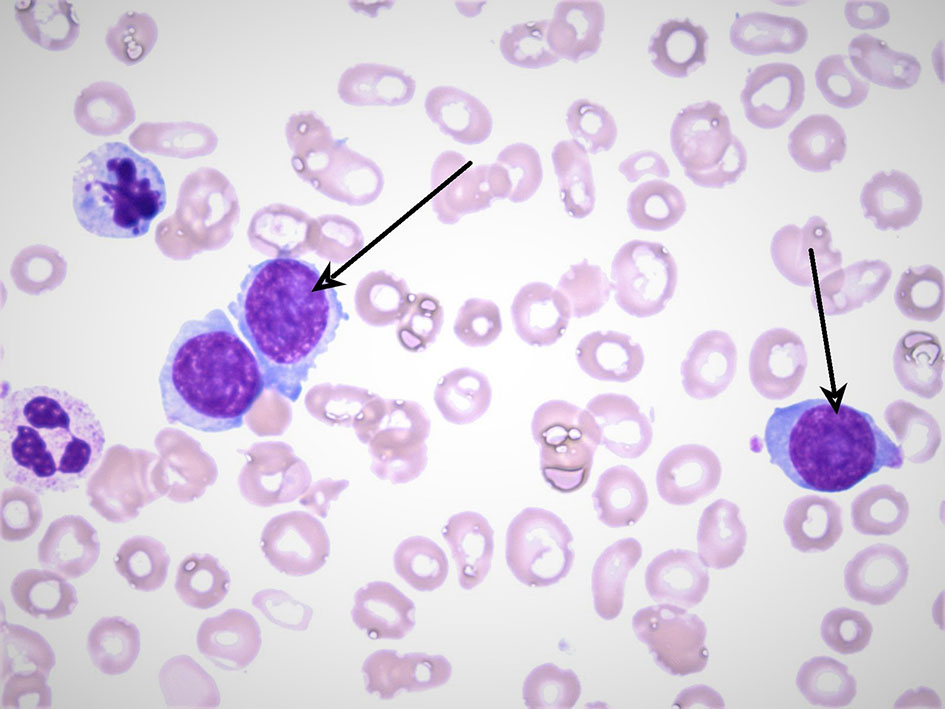 Click for large image | Figure 1. A peripheral blood smear with the arrows pointing at atypical plasma cells. The smear also reveals findings of anisopoikilocytosis and frequent target cells. Neutrophils are present with a mild left shift. Of note blasts are not seen. |
Chest X-ray revealed a large left pleural effusion. Computed tomography (CT) of the abdomen and pelvis was significant for retroperitoneal, periaortic, and aortocaval adenopathy, in addition to splenomegaly with a normal appearing liver. No lytic lesions were identified on any imaging studies.
Diagnostic and therapeutic left-sided thoracentesis was performed. The pleural fluid analysis was suggestive of a malignant exudative effusion: hazy, pink color, pH 8.0, fluid white blood cells 1,979 cells/µL (lymphocytes 23%, neutrophils 4%, plasma cells 52%, monocytes 21%), fluid red blood cells 14,528 µL, fluid total protein 2.6 g/L, and fluid LDH 198 U/L. Pleural involvement by plasma cell leukemia (PCL) was confirmed by the presence of plasma cells in the pleural fluid.
A hematologist consultation was requested for suspected plasma cell malignancy. Immunoglobulin levels revealed: immunoglobulin G (IgG) level of 4,400 mg/dL (normal range 700 - 1,600), IgA < 40 mg/dL (normal range 70 - 400), and IgM < 25 mg/dL (normal range 40 - 230). Bone marrow biopsy confirmed PCL with a hypercellular bone marrow consisting of CD138-positive kappa restricted plasma cells as seen in Figures 2, 3. CD20, CD56, CD117, and HLA-DR were negative. Cytogenetic analysis revealed high-risk abnormalities with deletion of 17p resulting in loss of p53, gain of 1q, 14q and t(11,14) translocation as displayed in Figure 2.
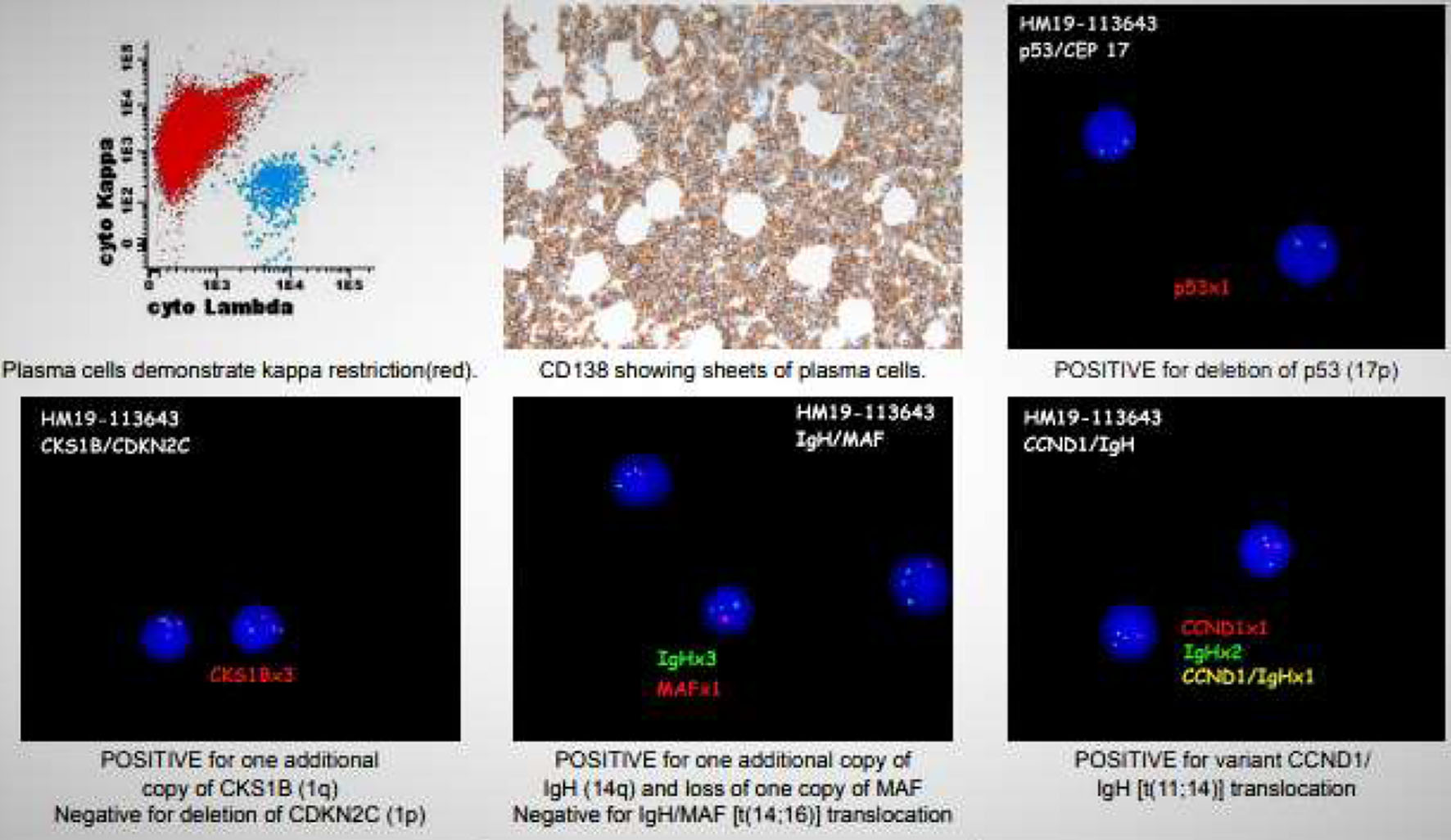 Click for large image | Figure 2. Multiple images including flow cytometry, bone marrow biopsy, and cytogenetic analysis (FISH). Flow cytometry (top left) shows a predominant kappa monotype restriction seen in red. Bone marrow biopsy (top middle) showing sheets of plasma cells 90-95%. FISH images display: a deletion of p53 (17p) (top right); an additional copy of CKS1B (1q) (bottom left); additional copy of IGH (14q) and loss of one MAF copy, no IGH/MAF t(14,16) translocation (bottom middle); positive variant CCND1/IGH t(11,14) translocation (bottom right). FISH: fluorescence in situ hybridization. |
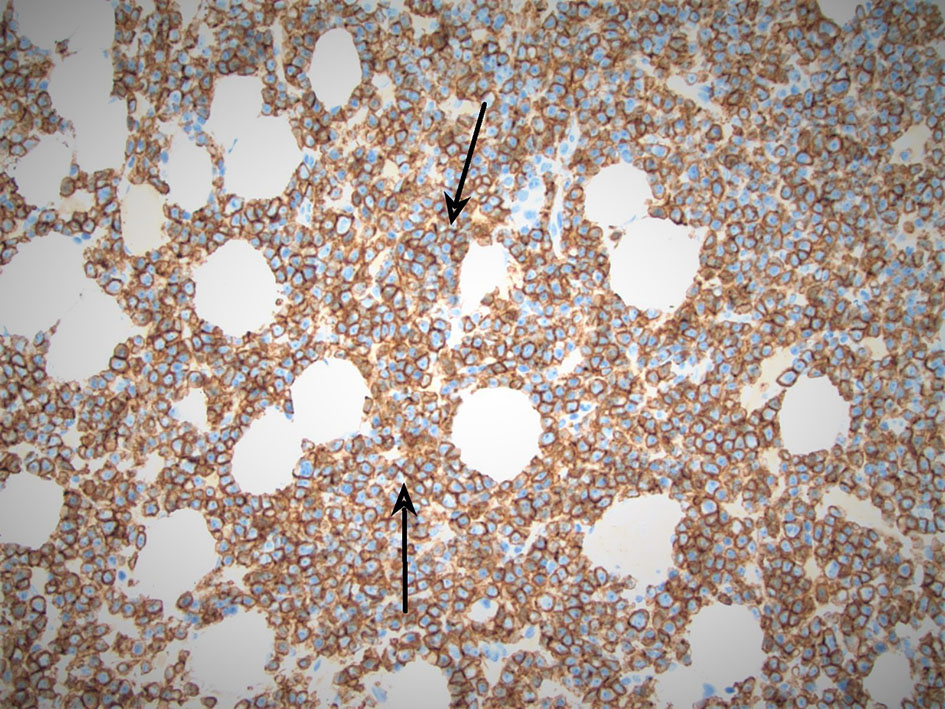 Click for large image | Figure 3. Arrows pointing at CD138-positive (kappa restricted) sheets of plasma cells, appearing in color blue. |
Lactulose administration led to improvement in mental status. Since no other etiology was identified, hyperammonemia was attributed to PCL. Central nervous system involvement was considered; however, CT scan of the head did not show any intracranial abnormalities on admission and further invasive testing such as lumbar puncture was not performed due to change in goals of care. She had rapid progression of her disease in the following 2 weeks with recurrent respiratory failure with re-accumulation of pleural effusion and dramatic decline in renal function. Induction chemotherapy was considered but due to her poor functional status, it was agreed upon that her overall condition precluded any cancer directed therapy. She was then transitioned to hospice.
| Discussion | ▴Top |
PCL is an aggressive plasma cell disorder which is a distinct clinicopathologic entity. It differs from MM, with a younger age at diagnosis, higher visceral and extramedullary involvement. The disease has unique biologic, immunophenotypic, and cytogenetic characteristics. PPCL is characterized by peripheral plasmacytosis detected at diagnosis not related to any underlying condition [2]. Secondary plasma cell leukemia (SPCL) is characterized by leukemic transformation after a diagnosis of MM has been made [3]. Historically around 60-70% of reported cases have been primary PCL, although secondary PCL is increasing as survival for MM has improved and now there appears to be an even distribution of primary versus secondary PCL [3, 4].
PCL is diagnosed using the Kyle’s criteria which consist of having at least 20% circulating plasma cells and a total plasma cell count of at least 2 × 109/L in the peripheral blood [2, 4, 5]. The most common laboratory findings include leukocytosis, anemia, and thrombocytopenia [3]. Osteolytic lesions are found in less than 50% of patients [3]. Extramedullary presentations such as hepatomegaly, splenomegaly, lymphadenomegaly, leptomeningeal infiltration or extramedullary plasmacytomas are more frequent in PPCL (23%) compared to MM (only 4%) [6]. PCL patients who present with high tumor burden typically have neoplastic cells that are monoclonal with either kappa or lambda light chain restriction. Light chain only PCL is more common than in MM, being the second most common subtype after the IgG subtype [7]. Bone marrow aspiration findings are similar to those seen in MM with increased number of monoclonal plasma cells [2]. There is no single cytogenetic abnormality that is typical or diagnostic of PCL. The most common abnormalities include deletion of chromosome 13 and monosomy 13. Del(17p), resulting in loss of TP53, have been detected in almost half of primary PCL and three-quarters of secondary PCL. In addition, PCL frequently has abnormalities in chromosome 1, in particular 1q21 amplification and del(1p21) [3]. Flow cytometry typically reveals expression of CD138 and CD38 [8, 9]. In contrast to most patients with MM, PCL frequently lacks CD56, CD117, HLA-DR expression but is more likely to express B cell marker CD20 [8, 9].
Historically prognosis was very poor with a median overall survival of 6 - 11 months for PPCL and 2 - 7 months for SPCL [3, 10]. With improvements in chemotherapy and the use of autologous stem cell transplantation, the median overall survival time has improved to 24 months for patients who were diagnosed during 2006 - 2009 compared to earlier decades, although first month mortality is still 15% [7, 11].
Current treatment for PCL is based on approved treatments for MM. Patients with good performance status are treated with induction regimens such as bortezomib, dexamethasone, thalidomide, cisplatin, doxorubicin, cyclophosphamide, and etoposide (VDT-PACE) followed by autologous hematopoietic cell transplantation (HCT) [12]. Older patients with poor functional status who are not candidates for HCT can be considered for bortezomib-based induction therapies such as bortezomib, thalidomide, dexamethasone (VTD), bortezomib, lenalidomide, dexamethasone (VRD), or bortezomib, cyclophosphamide, dexamethasone (VCD) [13, 14]. There are several ongoing trials evaluating second generation proteasome inhibitors carfilzomib and ixazomib as well as anti-CD38 and anti-CD45 antibodies [2, 15]. PCL carries a high risk of tumor lysis syndrome and treatment with rasburicase or allopurinol is recommended [12]. For PCL patients that relapse after induction therapy, maintenance therapy with lenalidomide and bortezomib rather than observation has been recommended [12].
Ammonia in humans is generated by bacterial hydrolysis of urea, other nitrogenous compounds in the intestine, the purine nucleotide cycle, amino acid transamination in skeletal muscle, and other metabolic processes [14]. If ammonia accumulates in the blood, it can cross the blood-brain barrier and result in neurological disorders such as disorientation, somnolence, confusion, and unconsciousness [14]. Non-hepatic causes of hyperammonemia can be seen in some hematologic malignancies after the administration of cytotoxic chemotherapy [16]. Although there are case reports of MM presenting with hyperammonemic encephalopathy, the reports of PCL presenting this way have not yet been published [17]. The pathophysiology of MM or PCL causing hyperammonemic encephalopathy remains unclear. Some studies have shown excess production of ammonia in vitro by human myeloma cell lines, but this is unclear if it is due to a mutation in ammonia production or whether it occurs due to the breakdown from excessive amino acids [18, 19]. Our patient did not have any objective evidence of hepatic dysfunction to explain hyperammonemia; PCL was considered the most likely explanation after other causes were excluded.
Conclusions
Non-hepatic hyperammonemia and subsequent encephalopathy can be a manifestation of PCL. PCL is an aggressive disease with poor survival. Treatment should be initiated without delay if clinical state and functional status permit. New treatment modalities are being investigated with hopes to improve overall survival in this fatal disease.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
The authors declare that it has been signed by the patient involved in the case.
Author Contributions
DP is the primary author and primary provider for the patient on admission to the hospital. He designed and developed main conceptual ideas and manuscript outline and wrote the first draft of the manuscript and made final critical edits. AM is a secondary author, reviewer, and has made significant contributions to editing. DM is a secondary author and primary provider for the patient on admission to the hospital. SE and KU are secondary care providers to the patient and have helped edit this paper. JH is the patients’ primary hematologist/oncologist and provided assistance with image procurement and interpretation. NB supervised the work, manuscript design and content, and provided critical edits to the final manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
PPCL: primary plasma cell leukemia; MM: multiple myeloma; LDH: lactate dehydrogenase; CT: computed tomography; SPCL: secondary plasma cell leukemia; MM: multiple myeloma; PCL: plasma cell leukemia; IgG: immunoglobulin G; VDT-PACE: bortezomib, dexamethasone, thalidomide, cisplatin, doxorubicin, cyclophosphamide, and etoposide; HCT: hematopoietic cell transplantation; VTD: bortezomib, thalidomide, dexamethasone; VRD: bortezomib, lenalidomide, dexamethasone; VCD: bortezomib, cyclophosphamide, dexamethasone; FISH: fluorescence in situ hybridization
| References | ▴Top |
- Winthrop P. Plasma Cell Leukemia. Rockefeller Cancer Institute. https://cancer.uams.edu/myeloma/myeloma-related-diseases/plasma-cell-leukemia.
- Gundesen MT, Lund T, Moeller HEH, Abildgaard N. Plasma cell leukemia: definition, presentation, and treatment. Curr Oncol Rep. 2019;21(1):8.
doi pubmed - Tiedemann RE, Gonzalez-Paz N, Kyle RA, Santana-Davila R, Price-Troska T, Van Wier SA, Chng WJ, et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia. 2008;22(5):1044-1052.
doi pubmed - Fernandez de Larrea C, Kyle RA, Durie BG, Ludwig H, Usmani S, Vesole DH, Hajek R, et al. Plasma cell leukemia: consensus statement on diagnostic requirements, response criteria and treatment recommendations by the International Myeloma Working Group. Leukemia. 2013;27(4):780-791.
doi pubmed - Kyle RA, Maldonado JE, Bayrd ED. Plasma cell leukemia. Report on 17 cases. Arch Intern Med. 1974;133(5):813-818.
doi pubmed - Igala M, Bopaka RG, Khtabi W, Benchekroun S, Quessar A. Primary plasma cell leukemia presenting as a thoracic mass. Pan Afr Med J. 2014;19:39.
doi pubmed - Gonsalves WI, Rajkumar SV, Go RS, Dispenzieri A, Gupta V, Singh PP, Buadi FK, et al. Trends in survival of patients with primary plasma cell leukemia: a population-based analysis. Blood. 2014;124(6):907-912.
doi pubmed - Craig FE, Foon KA. Flow cytometric immunophenotyping for hematologic neoplasms. Blood. 2008;111(8):3941-3967.
doi pubmed - Ioannou MG, Stathakis E, Lazaris AC, Papathomas T, Tsiambas E, Koukoulis GK. Immunohistochemical evaluation of 95 bone marrow reactive plasmacytoses. Pathol Oncol Res. 2009;15(1):25-29.
doi pubmed - Ngu S, Asti D, Valecha G, Thumallapally N, Pant M, Bershadskiy A. Primary plasma cell leukemia: A case report and review of the literature. Clin Case Rep. 2019;7(9):1702-1708.
doi pubmed - Ramsingh G, Mehan P, Luo J, Vij R, Morgensztern D. Primary plasma cell leukemia: a Surveillance, Epidemiology, and End Results database analysis between 1973 and 2004. Cancer. 2009;115(24):5734-5739.
doi pubmed - Rajkumar SV. Multiple myeloma: 2012 update on diagnosis, risk-stratification, and management. Am J Hematol. 2012;87(1):78-88.
doi pubmed - Pineda-Roman M, Zangari M, Haessler J, Anaissie E, Tricot G, van Rhee F, Crowley J, et al. Sustained complete remissions in multiple myeloma linked to bortezomib in total therapy 3: comparison with total therapy 2. Br J Haematol. 2008;140(6):625-634.
doi pubmed - Odigwe CC, Khatiwada B, Holbrook C, Ekeh IS, Uzoka C, Ikwu I, Upadhyay B. Noncirrhotic hyperammonemia causing relapsing altered mental status. Proc (Bayl Univ Med Cent). 2015;28(4):472-474.
doi pubmed - Pomalidomide, Ixazomib Citrate, and Dexamethasone in treating patients with previously treated multiple myeloma or plasma cell leukemia. Mayo Clinic. NCI. www.clinicaltrials.gov:NCT02547662.
- Mitchell RB, Wagner JE, Karp JE, Watson AJ, Brusilow SW, Przepiorka D, Storb R, et al. Syndrome of idiopathic hyperammonemia after high-dose chemotherapy: review of nine cases. Am J Med. 1988;85(5):662-667.
doi - Pham A, Reagan JL, Castillo JJ. Multiple myeloma-induced hyperammonemic encephalopathy: an entity associated with high in-patient mortality. Leuk Res. 2013;37(10):1229-1232.
doi pubmed - Murtaza G, Lu H, Faqah A, Konowitz N, Kuruvilla A, Adhikari S. Multiple myeloma-induced hyperammonemic encephalopathy. J Hematol. 2017;6(1):29-31.
doi pubmed - Eaton K, Holmstrom B, Abulhaija A. Hyperammonemic encephalopathy in multiple myeloma. Hospital Medicine. 2013;8(suppl 2).
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.