

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 12, Number 7, July 2021, pages 262-266
An Exceedingly Rare Case of Antithrombin III Deficiency and Catastrophic Antiphospholipid-Like Disease
Alexander T. Phana, b, c, Aldin Malkoca, b, Debra Craigb
aSt. George’s University School of Medicine, University Centre, Grenada
bArrowhead Regional Medical Center, 400 N Pepper Ave, Colton, CA 92324, USA
cCorresponding Author: Alexander Phan, St. George’s University School of Medicine, University Centre, Grenada
Manuscript submitted February 27, 2021, accepted March 13, 2021, published online May 13, 2021
Short title: AT III Deficiency and CAPS-Like Disease
doi: https://doi.org/10.14740/jmc3689
| Abstract | ▴Top |
Antithrombin III (AT III) is a critical component of the coagulation cascade that functions primarily to inhibit activated coagulation factors IIa and Xa. AT III deficiency is a disorder that predisposes patients to thromboemboli. Antiphospholipid syndrome (APS) is an autoimmune disorder that predisposes patients to vascular and microvascular thrombosis, which can often be devastating and lead to multiorgan involvement. The mainstay of treatment for both conditions involves the use of lifetime vitamin K antagonists. Recent studies suggest that patients with APS refractory to warfarin therapy may benefit from the addition of aspirin, statin, or hydroxychloroquine; low weight molecular heparin; or a combination regimen. Studies have also suggested that patients with AT III deficiency refractory to warfarin therapy may see improvement with use of a novel oral anticoagulant. This case report describes the recurrent hospitalizations of a 45-year-old patient who presented with multiorgan thrombosis involving the descending aorta, deep lower extremity veins, superior mesenteric artery and artery of the brain. This led to mesenteric ischemia, limb necrosis and a subacute frontal cortex infarct. Initial anticoagulation therapy was refractory to the use of warfarin. Enoxaparin therapy was initiated, resulting in no further thrombotic events. Clinicians should consider poor gastrointestinal absorption of warfarin in patients who fail to reach therapeutic anticoagulation goals. In addition, a thorough workup for hereditary and acquired thrombophilias should be performed in patients who present with recurrent thromboemboli, as these disorders increase the risk of poor patient outcomes if left untreated.
Keywords: Antithrombin; Heparin; Renal tubular acidosis; Warfarin; Enoxaparin; Venous thromboembolism
| Introduction | ▴Top |
Acquired coagulopathies are rare, and antithrombin III (AT III) deficiency is even more so due to the dearth of patient cases [1]. Normal AT III function provides most of its anticoagulation effects by inhibiting thrombin and other activated proteases of the coagulation system. AT III deficiency can present via congenital or acquired mechanisms. Various acquired causes may include decreased production and increased consumption, such as in liver disease, nephrotic kidney disease, or enteropathic protein loss. Most commonly, AT III deficiency leads to venous thromboemboli (VTE) of the deep veins and pulmonary vessels, leading to a seven-fold increased risk of VTE [2, 3]. As with most inherited thrombophilia syndromes, warfarin is an acceptable anticoagulant for VTE prevention; however, strict anticoagulation is required, which makes the treatment difficult for some patients.
Antiphospholipid syndrome (APS) is a systemic autoimmune condition diagnosed using the revised Sapporo classification, which includes vascular thrombosis or pregnancy loss. Laboratory diagnostic criteria include positivity for antiphospholipid antibodies measured by detection of lupus anticoagulant, beta-glycoprotein-I, and/or anticardiolipin antibody [4]. In the most severe form of APS, patients develop multiple organ thromboses recognized as catastrophic APS (CAPS) [5]. This study reports a unique case of a patient with both AT III deficiency and CAPS-like disease, characterized by multiorgan thromboses, low AT III activity, positive lupus anticoagulant and positive anti-cardiolipin. The patient developed these thrombotic events while anticoagulated with heparin and warfarin.
| Case Report | ▴Top |
Hospitalization 1
The patient in this case was a 45-year-old woman with past medical history of neurofibromatosis type I, renal tubular acidosis type II, coronary artery disease, peptic ulcer disease, non-diabetic gastroparesis and Takotsubo cardiomyopathy. She presented with left flank and generalized abdominal pain with radiation towards the back for 1 day. She also reported difficulty urinating and left lower extremity pain that worsened when walking. She denied fever, chills, cough, shortness of breath, chest pain, dysuria, hematuria, diarrhea, loss of sensation and weakness. She has a 15 pack-year tobacco smoking history, but denied alcohol consumption and illicit drug use. Physical examination revealed a 3/6 systolic murmur heard best at the right second intercostal space, absent bowel sounds, diffuse abdominal tenderness and absent pulses in the left foot.
Computed tomography (CT) of the abdomen revealed multiple mural thrombi of the descending thoracic and abdominal aorta, multiple wedge-shaped defects in the spleen and left kidney suggesting infarctions, inflammation indicating pancreatitis, distended gallbladder, bilateral non-obstructing renal stones, enlarged uterus and a mildly distended bladder. Figure 1a and b demonstrates splenic and hepatic infarcts, and a filling defect in the descending aorta. A CT angiogram of the lower extremities revealed a thrombus in the left common femoral artery causing 50% stenosis and occluded left posterior tibial and peroneal arteries proximally (Fig. 1c). Anti-cardiolipin immunoglobulin M (IgM) levels were also found to be elevated at 25 µg/L (normal ≤ 11 µg/L); however, repeat testing was negative. Protein S activity was noted to be low at 44% of normal despite being on heparin for anticoagulation. Testing for rheumatoid factor immunoglobulin G (IgG), anti-citrullinated peptide IgG, antinuclear antibody screen, ss-A antibody, ss-B antibody, double-stranded DNA antibody, beta-2 glycoprotein I (IgG, IgM and IgA), anti-cardiolipin (IgG and IgA), prothrombin G20210A mutation and factor V Leiden mutation were all negative. Protein C activity, activated protein C resistance and AT III levels were within normal limits.
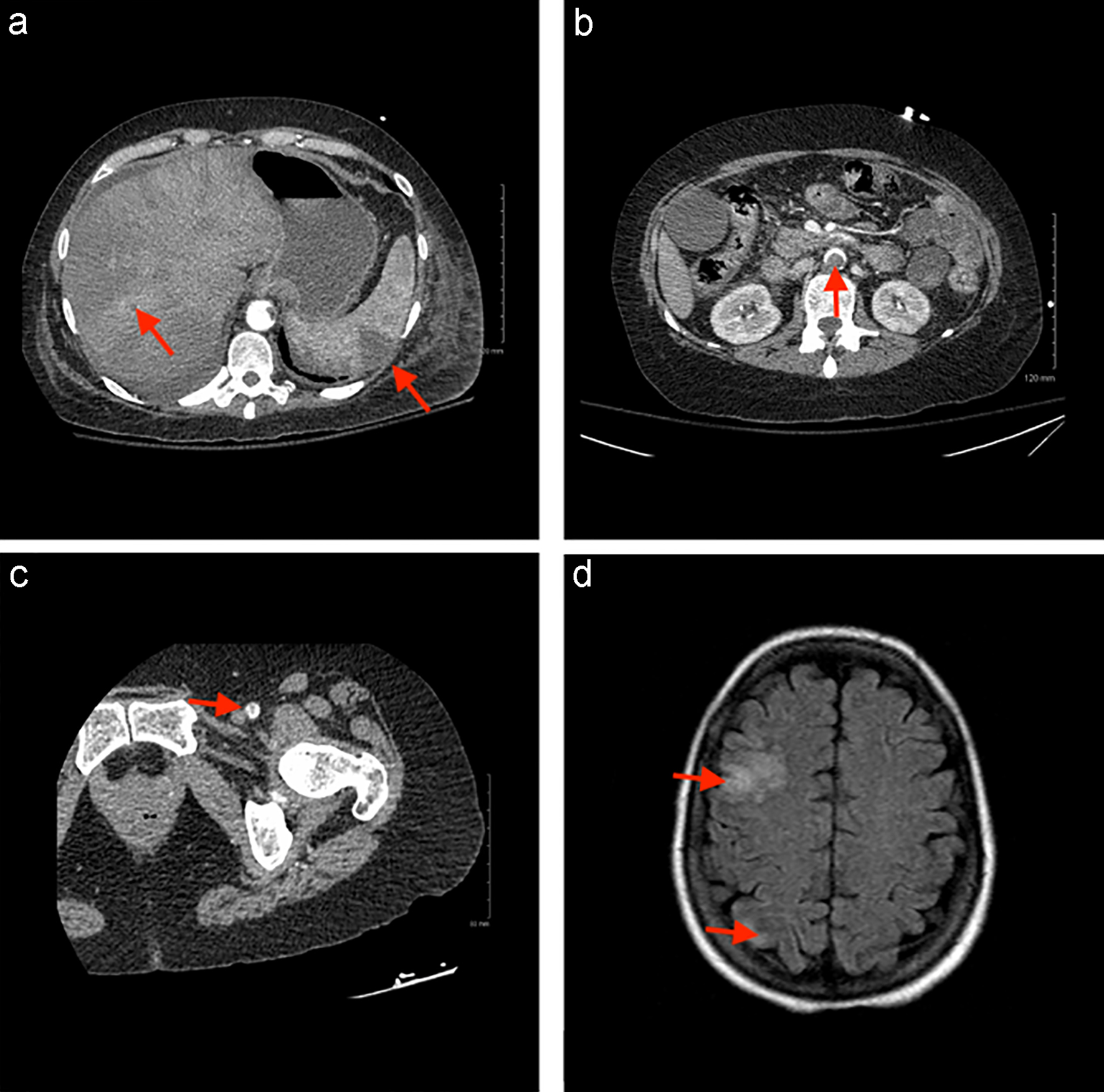 Click for large image | Figure 1. (a) Axial CT of the abdomen demonstrating splenic and hepatic infarcts. (b) Axial CT of the abdomen demonstrating filling defect in the descending aorta. (c) Axial CT of the left lower extremity demonstrating filling defect in the femoral artery. (d) Axial CT of the head demonstrating right frontal and occipital infarcts. CT: computed tomography. |
The next day, the patient became confused and had difficulty accessing her lexicon. A CT of the head showed a subacute small right frontal cortical infarct (Fig. 1d). She was subsequently transferred to the stroke unit for close neurologic observation. Her initial anticoagulation was managed with intravenous (IV) heparin (100 U/mL) for the first 15 days of hospitalization. On day 7, it was noted that she exhibited decreased protein S activity. The patient’s left lower limb second, third and fifth digits became progressively more cyanotic and darker in color throughout the stay. She was bridged to warfarin on day 16 and was maintained at an international normalized ratio (INR) goal of 2 - 3. On day 22, the patient tested positive for lupus anticoagulant. She was discharged on warfarin for anticoagulation management. Based on the clinical, radiologic and laboratory findings, the patient was diagnosed with CAPS-like disease.
Hospitalization 2
Six days later, she was sent to the hospital from the anticoagulation clinic due to multiple episodes of hematemesis, hematochezia and supratherapeutic INR. This was treated with vitamin K and prothrombin complex concentrate, which brought her INR down to 0.9 from > 10.8. An initial physical exam also revealed dry gangrene of the toes bilaterally. On admission, CT of the abdomen revealed an 11.5 cm peritoneal abscess in the lower abdomen and a new liver infarct. The abscess was incised and drained by interventional radiology on hospital day 4 and a Jackson-Pratt (JP) drain was left in place. She was administered IV heparin and bridged to warfarin on day 10. On day 29, the patient developed abdominal pain associated with intractable vomiting and nausea, which was concerning for ischemic bowel. A CT angiogram of the abdomen revealed a distal embolus of the superior mesenteric artery. Subsequent CT of the abdomen with contrast revealed a partial small bowel obstruction and a fistula between the cecum and abscess cavity.
On day 31, feculent and purulent substances were noted in the bulb of the JP drain. The patient continued to have remitting and relapsing episodes of abdominal pain, nausea and vomiting until day 46, when she noticed that her vomitus contained feculent material. An upper gastrointestinal and small bowel X-ray series with barium revealed a complete small bowel obstruction due to stricture of the terminal ileum. She was initially managed with decompression via nasogastric (NG) tube; however, the patient pulled out the NG tube the subsequent day. On day 47, laboratory testing revealed a low AT III activity of 60 (80 - 120), while on warfarin anticoagulation therapy. Due to the lab abnormalities, the patient was suspected to also have AT III deficiency.
On day 49, she was transitioned back to IV heparin. Due to malnutrition and poor tolerance of oral intake, she began total parenteral nutrition (TPN) via a peripherally inserted central catheter (PICC). To further characterize the fistula, a fistulogram was performed on day 52 and demonstrated an enteric fistula between the abscess cavity and terminal portion of the ileum with subsequent flow into the cecum. The primary team began to bridge her from heparin to warfarin, but her INR remained < 1.2, which never reached our therapeutic goal of 2 - 3. The patient was discharged on enoxaparin for anticoagulation management, scheduled for outpatient management of bilateral lower extremity digital necrosis and outpatient follow-up for colorectal surgery, and referred to a rheumatology clinic for further management of CAPS-like disease and AT III deficiency.
Hospitalization 3
Sixteen days later, the patient returned to the emergency department due to excruciating abdominal pain localized around the site of her JP drain. The patient reported that the pain started immediately after draining 700 mL of fluid from her drain. A CT of the abdomen revealed free fluid in the left paracolic gutter and the pelvis that appeared to be tracking from the abscess cavity, clinically correlating to her peritonitis. On day 2 of the third hospitalization, she was taken to the operating room and had an ileocecectomy with ileocolonic anastomosis performed.
The patient tolerated the procedure well and continued nutrition via TPN. Over the course of her hospitalization, she began to have regular bowel movements and progressively advanced to a regular oral diet. Her surgical wound healed well and her PICC line was removed on day 19. The patient’s anticoagulation was again managed with enoxaparin and she did not experience any thromboembolic events during this admission. She was discharged on enoxaparin for anticoagulation management, re-scheduled for outpatient management of bilateral lower extremity digital necrosis and outpatient management of CAPS-like disease and AT III deficiency.
Follow-up and outcome
At 1 month post-hospital discharge follow-up, the patient did not have any further thromboembolic events. However, 2 weeks after follow-up, the patient suddenly passed away. Due to this unfortunate outcome, we were unable to perform follow-up studies to meet the laboratory criteria for the revised Sapporo classification.
| Discussion | ▴Top |
Incidences of AT III deficiency and APS are extremely rare [1, 6]. AT III deficiency is diagnosed by AT III activity below the normal limit of 80 - 120. Differential diagnoses for decreased AT III activity include consumption, heparin therapy and nephrotic range proteinuria; however, our patient was on warfarin therapy for anticoagulation when her AT III activity was found to be low at 60 [7]. It should also be noted that our patient was never septic or diagnosed with disseminated intravascular coagulopathy. She was consequently diagnosed with acquired AT III deficiency.
APS diagnosis is based on the revised Sapporo classification criteria, as long as there is no alternative diagnosis. The revised Sapporo classification criteria are met when a patient presents with at least one clinical and one laboratory criteria (Table 1). CAPS is defined by: 1) Evident involvement of at least three organs, systems, or tissues; 2) Development of manifestations simultaneously or within 1 week; 3) Confirmation by histopathology of small vessel occlusion; and 4) Laboratory confirmation of antiphospholipid antibodies. However, if a patient does not meet all criteria, a combination of the aforementioned criteria results in a diagnosis of probable CAPS [4]. Although this patient does not meet all of the laboratory criteria for APS or probable CAPS, given her young age, multiple arterial thromboses, positivity for anticardiolipin IgM and positivity for lupus anticoagulant, she likely had CAPS-like disease.
 Click to view | Table 1. The Revised Sapporo Classification Criteria for Diagnosis of APS [4] |
AT III deficiency and APS are both typically managed with vitamin K antagonists [3, 8]. Although our patient did not meet the criteria for a definitive diagnosis of APS or CAPS, we managed her similarly to patients diagnosed with said conditions. In patients diagnosed with APS who fail to respond to warfarin therapy, adding low-dose aspirin, a statin, or hydroxychloroquine, low weight molecular heparin, or a combination of these regimens is recommended [9]. Despite initial warfarin therapy, this patient experienced worsening lower limb ischemia and an abdominal abscess secondary to mesenteric ischemia due to superior mesenteric artery thrombosis 6 days after initial discharge. Our patient likely failed to reach therapeutic INR with warfarin therapy due to poor gastrointestinal absorption and gastrointestinal complications. The decision was made to discharge the patient on enoxaparin for anticoagulation therapy. Despite her third admission to the hospital, continued enoxaparin therapy prevented recurrent thromboembolic events.
The primary limitation in this case is that our patient did not have subsequent lupus anticoagulant and anticardiolipin laboratory studies performed to meet the revised Sapporo classification criteria because she passed away. Although this limitation exists, given our patient’s clinical picture and lab findings, it is our contention that she had AT III deficiency and experienced CAPS-like disease. Recent literature suggests that long-term hydroxychloroquine therapy reduces rates of thrombotic events in patients with APS [9]. Although we did not attempt this treatment option, there is evidence that it may be a viable addition to standard APS treatment.
Learning points
In this patient, AT III deficiency and CAPS-like disease resulted in multiorgan insult, including the kidney, spleen, brain and gastrointestinal tract. Initial treatment of heparin bridge to warfarin was refractory, and due to poor absorption of warfarin, the patient required enoxaparin for anticoagulation therapy. New thrombosis ceased; however, previous thrombotic events resulted in gastrointestinal ischemia and dry gangrene of the patient’s toes. Clinicians should consider poor gastrointestinal absorption of warfarin in patients who fail to achieve adequate anticoagulation and consider a CAPS-like disease in patients who present with multiorgan thrombosis and antiphospholipid antibody positivity. This is a very serious patient presentation with a high likelihood of mortality.
Acknowledgments
The authors would like to thank and express their gratitude to Arrowhead Regional Medical Center’s exceptional nursing staff and unit managers for their expert clinical support.
Financial Disclosure
The authors have no financial or funding disclosures.
Conflict of Interest
The authors declare there is no conflict of interest.
Informed Consent
Informed consent was obtained from the family of the patient. The patient was also appropriately de-identified for this manuscript.
Author Contributions
AP contributed to the initial manuscript write-up, literature review and editing of the manuscript. AM contributed to the initial manuscript write-up, literature review and editing of the manuscript. DC attended on the case and contributed to decision-making, management of the patient, literature review and editing of the manuscript.
Data Availability
Any inquiries regarding supporting data availability of this study should be directed to the corresponding author.
Abbreviations
AT III: antithrombin III; APS: antiphospholipid syndrome; CAPS: catastrophic antiphospholipid syndrome; CT: computed tomography; DNA: deoxyribonucleic acid; IgG: immunoglobulin G; IgM: immunoglobulin M; IgA: immunoglobulin A
| References | ▴Top |
- Maclean PS, Tait RC. Hereditary and acquired antithrombin deficiency: epidemiology, pathogenesis and treatment options. Drugs. 2007;67(10):1429-1440.
doi pubmed - Kawano H, Maemura K. Edoxaban Was Effective for the treatment of deep vein thrombosis and pulmonary thromboembolism in a cancer patient with antithrombin III deficiency. Intern Med. 2016;55(22):3285-3289.
doi pubmed - Maeba H, Seno T, Shiojima I. Thrombectomy and catheter-directed thrombolysis combined with antithrombin concentrate for treatment of antithrombin deficiency complicated by acute deep vein thrombosis that is refractory to anticoagulation. Int Heart J. 2016;57(5):649-653.
doi pubmed - Aguiar CL, Erkan D. Catastrophic antiphospholipid syndrome: how to diagnose a rare but highly fatal disease. Ther Adv Musculoskelet Dis. 2013;5(6):305-314.
doi pubmed - Bucciarelli S, Espinosa G, Cervera R, Erkan D, Gomez-Puerta JA, Ramos-Casals M, Font J, et al. Mortality in the catastrophic antiphospholipid syndrome: causes of death and prognostic factors in a series of 250 patients. Arthritis Rheum. 2006;54(8):2568-2576.
doi pubmed - Miesbach W. Antiphospholipid antibodies and antiphospholipid syndrome in patients with malignancies: features, incidence, identification, and treatment. Semin Thromb Hemost. 2008;34(3):282-285.
doi pubmed - Heller EL, Paul L. Anticoagulation management in a patient with an acquired antithrombin III deficiency. J Extra Corpor Technol. 2001;33(4):245-248.
- Chighizola CB, Andreoli L, Gerosa M, Tincani A, Ruffatti A, Meroni PL. The treatment of anti-phospholipid syndrome: A comprehensive clinical approach. J Autoimmun. 2018;90:1-27.
doi pubmed - Garcia D, Erkan D. Diagnosis and management of the antiphospholipid syndrome. N Engl J Med. 2018;378(21):2010-2021.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.