

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 12, Number 10, October 2021, pages 400-404

Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal Protein, and Skin Changes Associated With Castleman Disease: A Common Misdiagnosis
Vanessa Palhaa, e ![]() , Elisa Condeza
, Elisa Condeza ![]() , Valentim Lopesb
, Valentim Lopesb ![]() , Carla Ferreirac
, Carla Ferreirac ![]() , Joana Sotto Mayora
, Joana Sotto Mayora ![]() , Teresa Pimentela
, Teresa Pimentela ![]() , Narciso Oliveiraa
, Narciso Oliveiraa ![]() , Marcio Tavaresd
, Marcio Tavaresd ![]()
aDepartment of Internal Medicine, Hospital de Braga, Braga, Portugal
bDepartment of Endocrinology, Hospital de Braga, Braga, Portugal
cDepartment of Pathology, Hospital de Braga, Braga, Portugal
dDepartment of Oncology, Hospital de Braga, Braga, Portugal
eCorresponding Author: Vanessa Palha, Department of Internal Medicine, Hospital de Braga, Sete Fontes-Sao Victor 4710-243, Braga, Portugal
Manuscript submitted July 22, 2021, accepted September 4, 2021, published online September 29, 2021
Short title: POEMS Syndrome Associated With CD
doi: https://doi.org/10.14740/jmc3759
| Abstract | ▴Top |
Castleman disease is a rare lymphoproliferative disorder. Co-presentation with polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes (POEMS syndrome) has been documented in 11-30% of Castleman disease cases. POEMS syndrome is a rare paraneoplastic disorder characterized by polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes. Not all features are required to make the diagnosis. We report a case of a woman who presented with a 1-year history of a left-side supraclavicular swelling associated with constitutional symptoms and symmetrical paresthesia of the lower limbs. In addition, she had skin hyperpigmentation, multiple supra and infra-diaphragmatic lymphadenopathies, hepatosplenomegaly and osteosclerotic lesions. Serum immunofixation was positive for immunoglobulin G-kappa gammopathy. A lymph node excisional biopsy was compatible with Castleman disease. The diagnosis of POEMS syndrome associated with Castleman disease was made. Our patient started treatment with a combination of bortezomib, cyclophosphamide, and dexamethasone with clinical and analytical improvement. Current treatment of POEMS syndrome associated with Castleman disease is focused on the management of POEMS syndrome. Early diagnosis requires a high index of suspicion and is crucial to reduce morbidity and mortality. This case report aims to raise awareness about this rare entity.
Keywords: Castleman disease; Monoclonal gammopathy; POEMS syndrome; Polyneuropathy
| Introduction | ▴Top |
Castleman disease (CD) is a rare [1, 2] and heterogeneous lymphoproliferative disorder [1-4], whose pathophysiology is not fully understood [1]. CD is classified clinically into unicentric or multicentric form (MCD) [1]. Co-presentation of MCD with polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes (POEMS syndrome) was first noted in the 1980 and 1990s [5], and it is estimated to occur in 11-30% of CD cases [1, 4].
POEMS syndrome is a rare paraneoplastic syndrome [3, 5, 6] that is described by the acronym in its name: polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes [2-8]. It was first described in 1938 when Scheinker reported a case of a 39-year-old man with sensorimotor peripheral neuropathy, solitary plasmacytoma and cutaneous hyperpigmentation [2, 4, 9]. The acronym was coined only in 1980 by Bardwick et al, and refers several but not all features of the POEMS syndrome [2, 3, 6, 7, 9]. Due to its rarity and wide clinical features, misdiagnosis as another disease is common [7, 10], and with significant morbidity and mortality.
Herein, we describe a case of a 69-year-old woman with a 1-year history of a left-side supraclavicular swelling associated with constitutional symptoms whose lymph node biopsy was compatible with CD. A review of symptoms and a complete physical examination followed by appropriate tests allowed an accurate diagnosis of POEMS syndrome associated with MCD. This case report aims to raise awareness about this entity.
| Case Report | ▴Top |
Investigations
We present a case of a 69-year-old woman, with a previous history of arterial hypertension and dyslipidemia. Her medication included amlodipine 5 mg, candesartan 8 mg, hydrochlorothiazide 12.5 mg and nebivolol 5 mg. She presented with a 1-year history of left-side supraclavicular swelling associated with progressive fatigue, anorexia, and weight loss (body weight: 74 to 63 kg). Cervical ultrasound documented multiple cervical lymphadenopathies with the largest lymph node (measuring 3.1 × 1.7 cm) being localized on the left supraclavicular region; and she was referred to the Department of General Surgery. A left supraclavicular lymph node excisional biopsy was performed. The histopathological examination of the specimen was consistent with the diagnosis of herpes virus-8-positive CD (HHV-8 CD, Fig. 1).
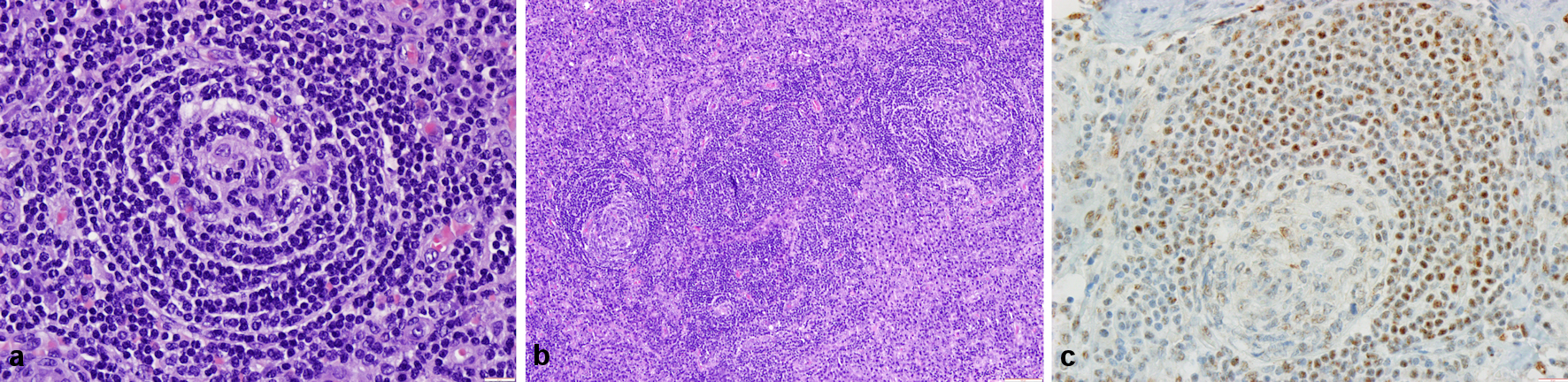 Click for large image | Figure 1. Histological findings in the surgical specimen. (a) A germinal center with lymphocyte depletion; small dark lymphocytes concentrically arranged in the mantle zone (hematoxylin and eosin (H&E), × 400). (b) Interfollicular regions expanded by sheets of plasma cells and vascular proliferation (H&E, × 100). (c) Immunohistochemical staining for human herpes virus-8 (HHV-8, × 400). |
She was then forwarded to the Department of Internal Medicine upon lymph node biopsy showed CD. Additional clinical history inquiry was attempted and revealed that, during the last few months, she noted progressive diffuse skin darkening and symmetrical paresthesia of the lower limbs. She denied fever, night sweats, gastrointestinal or respiratory complaints. Neurologic review of symptoms was negative for dysphagia, dysarthria, ocular changes, and bowel and bladder dysfunctions. She had no history of smoking, alcohol abuse, or illicit-drug use. She had no contact with animals or any sick person and did not have any recent travel outside Portugal. She had no family history of neurologic or oncologic diseases.
On physical examination, she was afebrile. Her blood pressure was 151/77 mm Hg and heart rate 62 beats/min, and peripheral oxygen saturation was 97% while on room air. She had diffuse cutaneous hyperpigmentation and palpable lymph nodes in her cervical and supraclavicular regions, with the largest measuring 3 × 2 cm. Abdominal examination revealed an enlarged liver and no other abnormality. She had bilateral edema of lower limbs. Neurological examination revealed hypoesthesia below the ankles and tendon hyporreflexia; her muscle strength was decreased in upper and lower extremities with muscle strength grade 4-/5 proximal and grade 4+/5 distal.
Diagnosis
Blood tests showed normal complete blood count, coagulation tests, serum bilirubin (total and conjugated), aspartate and alanine aminotransferases and lactate dehydrogenase. Erythrocyte sedimentation rate and C-reactive protein were within normal range. Renal function, serum calcium and total serum protein were normal. Endocrine screening showed normal thyroid function, a normal glycemic profile (fasting plasma glucose and hemoglobin A1c), and a normal cortisol dosage. Hepatitis B and C, toxoplasmosis, tuberculosis, human immunodeficiency virus (HIV), cytomegalovirus (CMV) and Epstein-Barr virus (EBV) infections were ruled out. Auto-immune screening (antinuclear antibodies (ANA), anti-double-stranded deoxyribonucleic acid (dsDNA) and rheumatoid factor) was negative with no complement consumption. Albumin was 3.2 g/dL (reference range (RR): 3.4 - 5) and serum protein electrophoresis (SPEP) with immunofixation demonstrated a monoclonal immunoglobulin G (IgG)/kappa peak with IgG level of 2,120 mg/dL (RR: 650 - 1,600). Serum free light chains ratio was normal. Bence-Jones proteins were not detected. Bone marrow biopsy showed the presence of reactive-appearing lymphocytes B and T and 5% of plasma cells (Fig. 2).
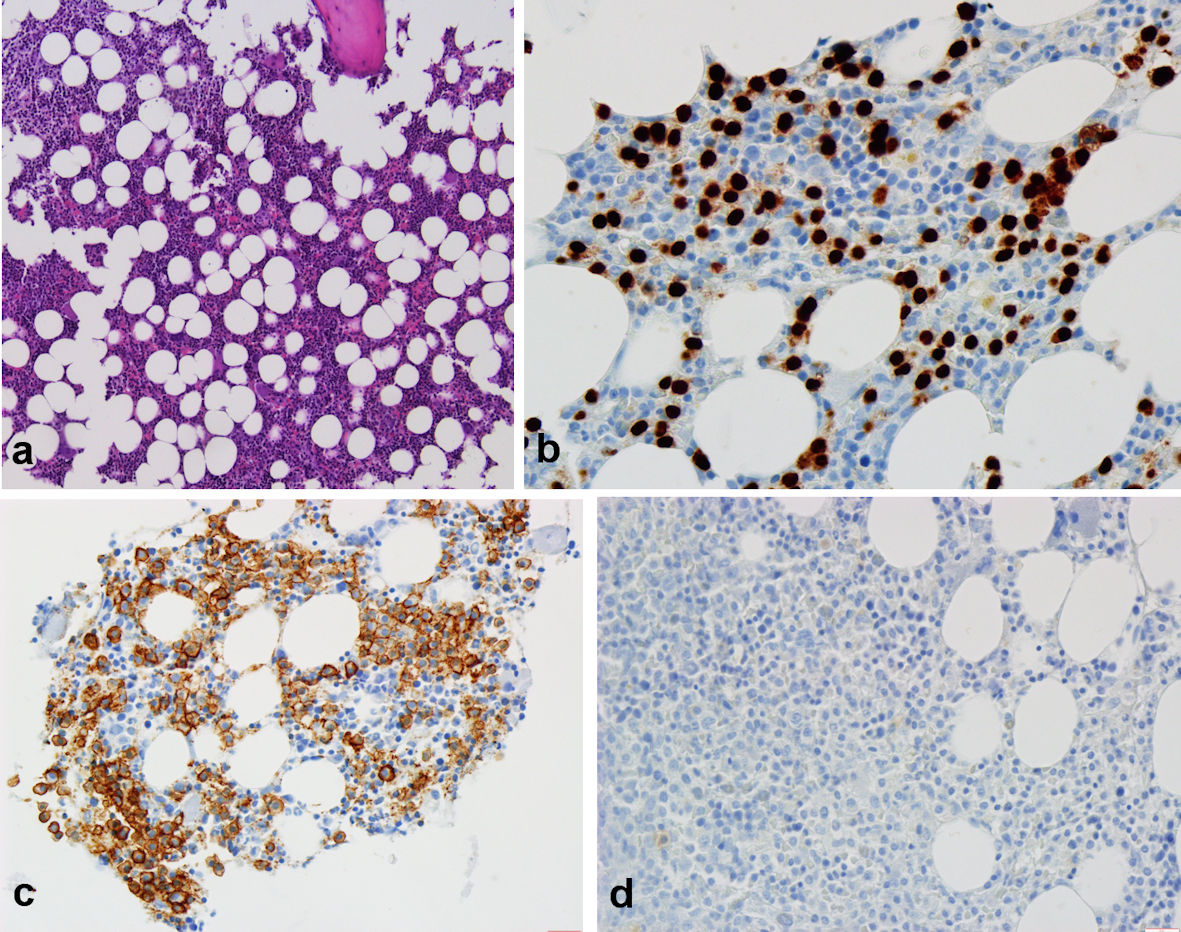 Click for large image | Figure 2. Histological findings in the bone marrow biopsy specimen. (a) Elements from the three hematopoietic lineages with normal density, location and maturation (hematoxylin and eosin (H&E), × 100). (b, c) Positive immunohistochemical staining for plasma cells accounting for about 5% of the total cell population ((b) CD138, × 400); (c) MUM1, × 400). (d) Negative immunohistochemical staining for human herpes virus-8 (HHV-8, × 400). |
Plasma vascular endothelial growth factor (VEGF) measurement was elevated (644 pg/mL, RR: < 115). Human herpes virus-8 (HHV8) was not detected in peripheral blood by polymerase chain reaction.
Thoraco-abdominopelvic computed tomography scan revealed small bilateral pleural effusion, multiple enlarged supra and infra-diaphragmatic lymphadenopathies, hepatosplenomegaly and multiple osteosclerotic lesions (Fig. 3). On transthoracic echocardiography there was a normal systolic function without pulmonary hypertension or pericardial effusion. Electromyography was consistent with sensorimotor peripheral polyneuropathy.
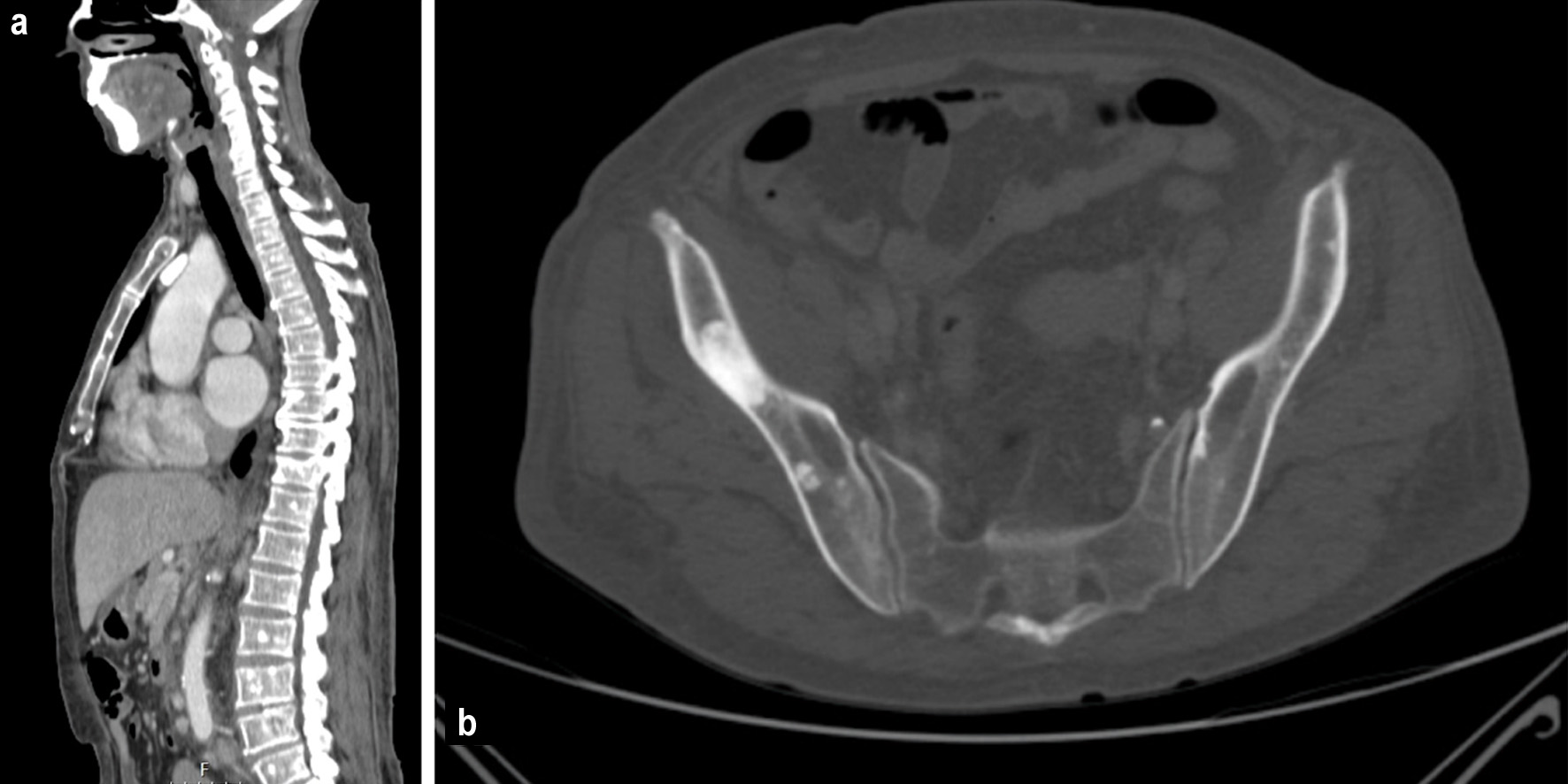 Click for large image | Figure 3. Bone windows of computed tomography scan showing: (a) diffuse sclerotic lesions and (b) sclerotic lesion in right ischium. |
Thus, our patient meets the criteria for the diagnosis of POEMS syndrome in association with CD.
Treatment
She was referred to the Hematology Department, and she started treatment with a combination of bortezomib, cyclophosphamide, and dexamethasone.
Follow-up and outcomes
After the first cycle, she noticed improvement of paresthesia and IgG level reduced from 2,120 to 960 mg/dL (RR: 650 - 1,600). She has tolerated treatment well without any clinically significant adverse effect.
| Discussion | ▴Top |
CD, also known as angiofollicular lymph node hyperplasia, was first described in 1956 by Castleman et al [1, 8], and is a rare lymphoproliferative disorder [1, 4, 8]. CD is clinically classified into unicentric when only a single enlarged lymph node or region of lymph nodes is involved, or multicentric (MCD) disease, which involves multiple lymph node regions [1].
Once biopsy suggests CD, further investigation should be focused on excluding diseases with CD-like adenopathy and identifying associated conditions. Even if a lymph node biopsy was consistent with CD, multiple diseases have been associated with Castleman-like histopathologic features such as autoimmune disorders like rheumatoid arthritis or systemic lupus erythematosus [5]. Nevertheless, in our patient, history and physical examination were not compatible and serologic tests excluded those diagnoses. Infectious diseases such as HIV, tuberculosis, CMV and EBV virus infection were excluded too.
The findings on neurologic examination made us consider possible associated conditions. The combination of polyneuropathy associated with a monoclonal protein is seen in monoclonal gammopathy of undetermined significance (MGUS), multiple myeloma (MM) [4, 6], Waldenstrom macroglobulinemia (WM) [6], amyloidosis, CD, and POEMS syndrome [4, 6]. Despite the presence of a monoclonal peak on SPEP, the diagnosis of MM seemed unlikely given the absence of osteolytic lesions, anemia, hypercalcemia, and renal failure. Furthermore, bone marrow showed less than 10% of clonal plasma cells and polyneuropathy is uncommon in classic MM. MGUS occurs in the absence of systemic findings, beyond plasma cell proliferative disorder. Like MM, the age onset of MGUS is typically older than for POEMS syndrome. WM may be complicated by lymphadenopathies, organomegalies and polyneuropathy; however, WM is classically associated with IgM monoclonal gammopathy and the presence of abnormal lymphoplasmacytic cells in bone marrow examination [6].
Several authors have reported a close association between MCD and POEMS syndrome [1, 4, 5]. In a study with 113 patients with MCD, 32% met the criteria for the POEMS syndrome diagnosis [8], it should always be ruled out since it changes the patient’s management.
POEMS syndrome, also known as osteosclerotic myeloma, Crow-Fukase syndrome or Takatsuki syndrome [3, 7], is a rare multisystemic disorder [4, 6, 7], mainly affecting men in the fifth and sixth decades of life [4, 6, 9]. Peripheral polyneuropathy and monoclonal plasma cell disorder are the two cardinal features, and when both are present, the diagnosis should be suspected.
According to the International Myeloma Working Group criteria, the diagnosis of POEMS syndrome requires the presence of both mandatory criteria (polyneuropathy and monoclonal plasma cell proliferative disorder), plus at least one major criterion (sclerotic bone lesions, CD and elevated VEGF), and at least one minor criteria (organomegaly, extravascular volume overload, endocrinopathy, skin changes, papilledema and thrombocytosis or polycythemia) [2, 3, 6].
The clinical features of POEMS syndrome widely differ among patients and all symptoms are not always present [3, 6]. Patients may experience a delay in the diagnosis [7, 8] with a median time from onset to diagnosis of 18 months [10]. Prompt diagnosis is critical to reduce the morbidity rate and increase survival.
Our patient meets criteria for POEMS syndrome based on polyneuropathy, monoclonal plasma cell disorder, sclerotic bone lesions, CD, elevated VEGF, hepatosplenomegaly, hyperpigmentation, and extravascular overload. Additionally, weight loss is also present, and it is known to be associated [6, 9]. Thus, our patient was diagnosed with POEMS syndrome associated with CD.
Interestingly, the type of light chain found in POEMS syndrome is almost always lambda [4-6, 9], although kappa light chain could also be seen. It is not known if there is any clinical manifestation difference regarding the involved light chain.
The pathogenesis of POEMS syndrome remains unclear [2-4, 7], but VEGF associated with interleukin (IL)-6 and IL-12 seems to play a crucial role [2, 4, 7]. An elevated plasma VEGF supports the diagnosis [3, 4, 6], and could be used to monitor disease activity [2-4, 6].
Treatment for the MCD associated with POEMS syndrome is focused on the management of the plasma cell proliferative disorder. Due to its rarity, treatment for the POEMS syndrome is not standardized [2, 3]; and recommendations are based on limited trial data, case series and anecdotal data [3, 7]. For patients with localized disease, defined as the presence of an isolated bone disease without clonal plasma cells on bone marrow biopsy, radiation therapy is potentially curative [3]. Patients with disseminated bone marrow involvement, require treatment with standard MM therapy: high-dose chemotherapy with autologous stem cell transplant [2, 3, 6, 7]. If treated appropriately, the prognosis of POEMS syndrome is good [2, 6] with large series reporting average survival around 13.7 years [9]. POEMS syndrome associated with MCD is rarely seen and more reports are needed to understand its natural history.
Learning points
In patients with lymph node biopsy compatible with CD, POEMS syndrome should be sought after with appropriate clinical, laboratory, and radiographic workup. POEMS syndrome should be considered in the presence of peripheral polyneuropathy and monoclonal plasma cell disorder. Early diagnosis of POEMS syndrome requires a high index of suspicion and is crucial to reduce morbidity and mortality.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Informed consent was obtained.
Author Contributions
VP was responsible for conception and writing the manuscript. EC, VL, TP, NO and JSM contributed to performing the diagnostic investigation and data analysis. MT was responsible for the patient’s hematological evaluation and participated in the diagnostic investigation. CF was responsible for the lymph node and bone marrow biopsy reports and images. All authors revised critically the work for important intellectual content. All authors approved the final manuscript and are accountable for all aspects of the work.
Data Availability
Any inquiries regarding supporting data availability of this study should be directed to the corresponding author.
Abbreviations
CD: Castleman disease; CMV: cytomegalovirus; EBV: Epstein-Barr virus; HHV8: human herpes virus-8; HIV: human immunodeficiency virus; IL: interleukin; MCD: multicentric Castleman disease; MGUS: monoclonal gammopathy of undetermined significance; MM: multiple myeloma; WM: Waldenstrom macroglobulinemia; SPEP: serum protein electrophoresis; VEGF: vascular endothelial growth factor
| References | ▴Top |
- Haap M, Wiefels J, Horger M, Hoyer A, Mussig K. Clinical, laboratory and imaging findings in Castleman's disease - The subtype decides. Blood Rev. 2018;32(3):225-234.
doi pubmed - Ali T, Qazilbash MH. POEMS syndrome: A multisystem clonal disorder. Eur J Haematol. 2021;106(1):14-18.
doi pubmed - Dispenzieri A. POEMS Syndrome: 2019 Update on diagnosis, risk-stratification, and management. Am J Hematol. 2019;94(7):812-827.
doi pubmed - Dispenzieri A, Kourelis T, Buadi F. POEMS Syndrome: Diagnosis and Investigative Work-up. Hematol Oncol Clin North Am. 2018;32(1):119-139.
doi pubmed - Dispenzieri A, Fajgenbaum DC. Overview of Castleman disease. Blood. 2020;135(16):1353-1364.
doi pubmed - Brown R, Ginsberg L. POEMS syndrome: clinical update. J Neurol. 2019;266(1):268-277.
doi pubmed - Shi H, Jiang X, Wang L, Zhou J. Missed diagnosis of POEMS syndrome with onset of progressive fatigue and numbness: a case report. J Int Med Res. 2021;49(2):300060520986706.
doi pubmed - Kang J, Yang F, Zhang HY, Hu MM, Xia F, Wang JC, Deng YC, et al. POEMS syndrome associated with Castleman disease: a case report and literature review. Neuroimmunol Neuroinflammation. 2014;1(1):40-43.
doi - Marinho FS, Pirmez R, Nogueira R, Cuzzi T, Sodre CT, Ramos-e-Silva M. Cutaneous manifestations in POEMS syndrome: case report and review. Case Rep Dermatol. 2015;7(1):61-69.
doi pubmed - Li J, Zhou DB, Huang Z, Jiao L, Duan MH, Zhang W, Zhao YQ, et al. Clinical characteristics and long-term outcome of patients with POEMS syndrome in China. Ann Hematol. 2011;90(7):819-826.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.