

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 13, Number 5, May 2022, pages 235-239

Idiopathic Pleuroparenchymal Fibroelastosis
Kameron Tavakoliana, c, Ndausung Udongwoa, Steven Douedia, Mihir Odaka, Justin Ilagana, Taimoor Khanb, Noor Salamb, Saira Chaughtaia, Arif Asifa
aDepartment of Medicine, Jersey Shore University Medical Center, Neptune City, NJ, USA
bDepartment of Pulmonary and Critical Care, Jersey Shore University Medical Center, Neptune City, NJ, USA
cCorresponding Author: Kameron Tavakolian, Department of Internal Medicine, Jersey Shore University Medical Center, Neptune City, NJ 077543, USA
Manuscript submitted March 28, 2022, accepted April 21, 2022, published online May 7, 2022
Short title: A Case of IPPFE
doi: https://doi.org/10.14740/jmc3927
| Abstract | ▴Top |
Idiopathic pleuroparenchymal fibroelastosis (IPPFE) is a rare form of idiopathic interstitial pneumonia. The disease is characterized by fibrosis of the pleura and subpleural lung parenchyma predominantly affecting the upper lobes. Various triggers have been proposed as inciting factors in the development of the disease. Diagnosis is made clinically in conjunction with radiographic findings and histopathology when available. There are no known effective treatment options and several cases of lung transplantation have been reported. We report a case of an 86-year-old female who presented to the emergency department with worsening dyspnea and hypoxia. She had a history of unexplained pneumomediastinum and a 20 - 25 pounds unintentional weight loss over 10 months. Computed tomography (CT) of the chest without contrast revealed radiographic evidence of IPPFE. Despite symptomatic management with antibiotics, diuretics, and steroids, her condition continued to deteriorate. Unfortunately, our patient was not a candidate for a lung transplant. She was transitioned to hospice care and succumbed to her disease. IPPFE is a rare disease with an unknown prevalence. It has a median survival rate of 2 years. Usually, there is an overlap with interstitial lung diseases, making it challenging to diagnose. There are only a few cases reported in the literature, and there are currently no guidelines available on the appropriate management of this debilitating disease. We recommend more cases be reported, and further research is done to establish better criteria for diagnosis and management.
Keywords: Idiopathic pleuroparenchymal fibroelastosis; Interstitial lung disease; Interstitial pneumonia; Platythorax; Pneumothorax; Pneumomediastinum; Dyspnea; Hypoxia
| Introduction | ▴Top |
Idiopathic pleuroparenchymal fibroelastosis (IPPFE) is a rare condition that was first described in Japanese literature in 1992 by Amitani et al as idiopathic upper lobe pulmonary fibrosis [1]. In 2013, the condition was recognized as a rare idiopathic interstitial pneumonia by the American Thoracic Society/European Respiratory Society. The disease is characterized by fibrosis of the pleura and subpleural lung parenchyma that predominantly affects the upper lobes [2]. Histopathology demonstrates intra-alveolar fibrosis and elastosis with visceral pleural fibrosis [3]. The pathogenesis of the disease is not fully understood, but proposed triggers include lung or bone marrow transplant, chemotherapy treatment, connective tissue disease, recurrent pulmonary infections, and occupation dust inhalation [4-6]. We present a case of an 86-year-old female who was clinically diagnosed with IPPFE after presenting with typical symptoms and characteristic radiographic findings seen in IPPFE.
| Case Report | ▴Top |
Investigations
An 86-year-old female with a history of breast cancer status post lumpectomy and radiation on the right, gastroesophageal reflux disease, chronic obstructive pulmonary disease, hypertension, osteoporosis, anxiety, chronic back pain, and recent admission 6 months prior for pneumomediastinum of unclear etiology presented to the emergency department with worsening dyspnea and hypoxia. Symptoms started about 1 month prior to her recent admission. It was associated with intermittent episodes of productive cough (non-bloody sputum), constipation, decreased appetite, fatigue, and unintentional weight loss of 20 - 25 pounds within 10 months. She denied any fever, chills, chest pain, nausea, vomiting, recent travel history, or sick contacts. Her family history was remarkable for lung cancer in her father (unspecified age). Of note, she received three doses of Pfizer severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) vaccine. The patient had not traveled outside the United States for over 20 years, did not have any known occupational or environmental exposures, and denied any history of smoking or recurrent pulmonary infections. Her home medications were tramadol 50 mg every 6 h as needed (PRN), alprazolam 0.5 mg three times daily PRN, buspirone 15 mg twice daily, hydrochlorothiazide 25 mg daily, levothyroxine 50 mg daily, calcitonin 200 U/act one nasal spray daily, megestrol 400 mg daily, and montelukast 10 mg daily. On initial evaluation, vitals were temperature 36.3 °C, heart rate 99 beats per minute, blood pressure 134/79 mm Hg, and saturating 88% on room air. She was subsequently placed on 4 L of supplemental oxygen via nasal cannula and her oxygen saturation improved to 95%. Her body mass index (BMI) was 14.4 kg/m2 (normal value: 18.5 - 24.9 kg/m2). The physical exam was notable for a frail elderly lady with a prominent suprasternal notch and bilateral inspiratory crackles. Cardiovascular examination was unremarkable, with no jugular venous distention, murmurs, rubs, or gallops. Also, there was no lower extremity edema.
Diagnosis
Laboratory studies showed sodium of 137 mmol/L (normal value: 135 - 146 mmol/L), potassium of 3.3 mmol/L (normal value: 3.5 - 5.0 mmol/L), chloride of 96 mmol/L (normal value: 96 - 110 mmol/L), bicarbonate of 32 mmol/L (normal value: 24 - 31 mmol/L), alkaline phosphatase of 169 U/L (normal value: 38 - 126 U/L), total protein of 5.7 g/dL (6.0 - 8.0 g/dL), albumin of 2.6 g/dL (normal value: 3.5 - 5.0 g/dL), aspartate aminotransferase of 91 U/L (normal value: 10 - 42 U/L), alanine aminotransferase of 97 U/L (normal value: 10 - 60 U/L), platelet count of 124 × 103/µL (normal value: 140 - 450 × 103/µL), and procalcitonin 0.33 ng/mL (normal value: < 0.50 ng/mL) as shown in Table 1. Additional relevant negative workup included Streptococcus pneumoniae (urine antigen), Legionella (urine antigen), respiratory pathogen panel, QuantiFERON-TB, and 1,3-β-d-glucan, also shown in Table 1.
 Click to view | Table 1. Initial Laboratory Results |
Electrocardiogram showed normal sinus rhythm with a rate of 85 beats per minute, left axis deviation with T wave inversions in leads III, aVF, V3 - V6. Chest X-ray (CXR) posterior-anterior and lateral views showed mild cardiomegaly, biapical pleural thickening, minimal pulmonary vascular congestion superimposed on chronic changes, and small bilateral pleural effusions (Fig. 1). Computed tomography (CT) scan of the chest revealed upper lobe predominant interstitial lung disease with thickened pleura (Fig. 2), as well as traction bronchiectasis, basilar reticulations, and honeycombing (Fig. 3). Autoimmune serological testing and extensive infectious workup was negative. At this point, the leading differential diagnoses included IPPFE, hypersensitive pneumonitis, radiation pneumonitis, acute fibrinous and organizing pneumonia, and idiopathic bronchiolocentric interstitial pneumonia. The patient’s history, coupled with her clinical presentation and radiographic imaging findings placed IPPFE as the culprit for the worsening dyspnea.
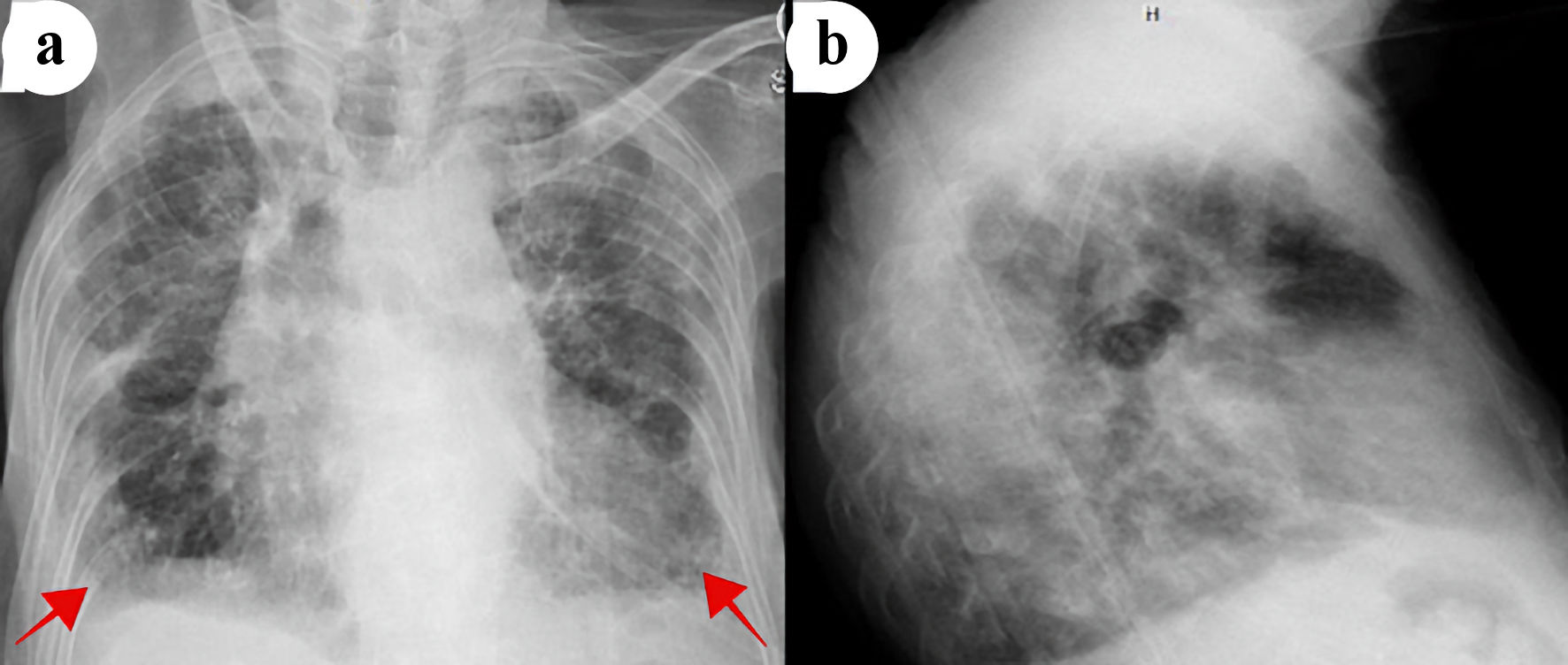 Click for large image | Figure 1. Chest X-ray posterior-anterior (a) and lateral (b) views showing cardiomegaly, pulmonary vascular congestions, and small bilateral pleural effusions (red arrows). |
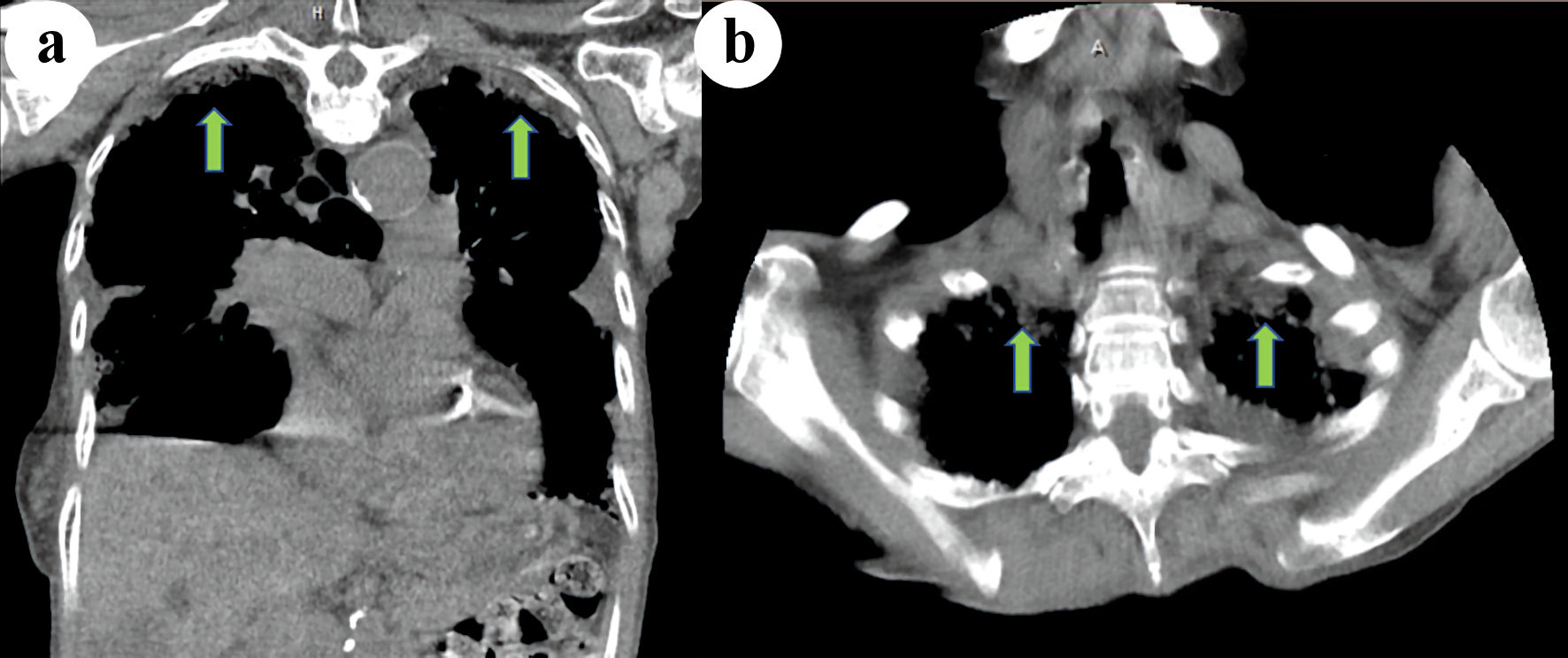 Click for large image | Figure 2. Coronal (a) and axial (b) computed tomography (CT) scan of the chest demonstrating bilateral apical pleural thickening (green arrows). |
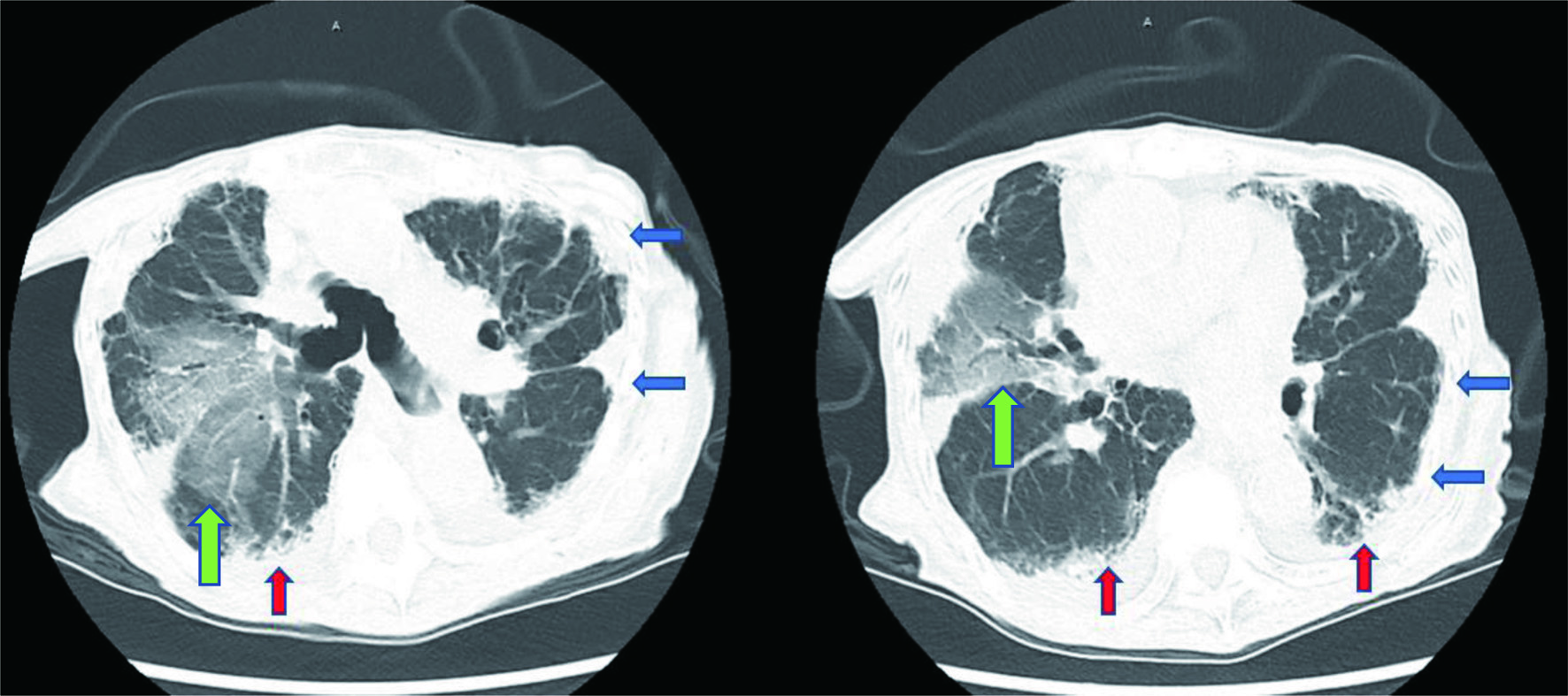 Click for large image | Figure 3. Computed tomography (CT) scan of the chest showing upper lobe predominant interstitial lung disease with thickened pleura (blue arrows), as well as traction bronchiectasis, basilar reticulations, and honeycombing (red arrows). Right lung opacities (green arrows) are observed in the location of the prior radiation treatment. |
Treatment
The patient was treated with doxycycline 100 mg every 12 h for 7 days and prednisone 40 mg daily for 5 days. Nine days following her admission, discharge to rehab center was canceled after a rapid response was called for hypoxia and respiratory distress. Repeat CXR showed no significant changes when compared to prior. She received 20 mg of intravenous (IV) furosemide and 125 mg of IV methylprednisolone. However, her condition continued to deteriorate, requiring an increasing amount of supplemental oxygen. The patient was ultimately transferred to the intensive care unit (ICU) for closer monitoring. While in the ICU, there were daily evaluations made by the cardiology, pulmonary, and gastrointestinal team for demand-supply mismatch, dyspnea/hypoxia, and gastrointestinal bleed respectively. Alternating use of bi-level positive airway pressure (BiPAP) and high-flow oxygen was used in the management of hypoxia. The patient and family declined intubation with mechanical ventilation after meeting with the care team. Furthermore, stress dose steroids did not improve symptoms.
Follow-up and outcomes
Due to the patient’s respiratory failure becoming refractory to available therapies, a lung biopsy to elucidate the cause of respiratory failure was deferred, and comfort care measures were agreed upon by her family and the treatment team. The patient was transitioned to hospice care and succumbed to her disease shortly after. Attempts were made to obtain post-mortem tissue sample for definitive confirmation of IPPFE; however, family felt this was futile.
| Discussion | ▴Top |
IPPFE is a rare form of idiopathic interstitial pneumonia with an unknown prevalence [4]. The pathogenesis is thought to involve diffuse alveolar damage leading to interstitial inflammation and subsequent fibrosis [7]. Proposed inciting triggers for the development of IPPFE include lung, bone marrow, and stem cell transplantation, fibrotic interstitial lung disease, recurrent pulmonary infection, autoimmune disease, and environmental exposures to asbestos or aluminum. In addition, mutations in genes involving telomere function have been linked to a more severe phenotype of the disease [8].
A review of 78 cases reported a bimodal age distribution ranging between 13 and 85 years of age, with a mean of 49 years. There is mixed evidence regarding gender preference; however, one recent study found that patients with IPPFE were significantly more likely to be non-smoking females with low BMI [9].
The most common presenting symptoms are exertional dyspnea, dry cough, weight loss, and chest discomfort. Additionally, spontaneous pneumothorax and pneumomediastinum have been reported [10]. Clinical exam features include inspiratory crackles on auscultation, platythorax (decreased anteroposterior thoracic depth and flattening of the frontal chest), and deepened suprasternal notch [4]. One study that assessed average anteroposterior thoracic depth in adults based on CT scan imaging found the average female depth to be 235 ± 30 mm [11]. The characteristic finding on a high-resolution CT (HRCT) scan is pleural thickening with subpleural fibrosis localized to the upper lobes [3]. Although not required for diagnosis, histopathology demonstrates intra-alveolar fibrosis and elastosis with visceral pleural fibrosis [3]. Establishing the diagnosis of IPPFE is controversial. The process involves clinical, radiological, and if available, histopathological information. Surgical biopsy is commonly avoided due to the risk of developing pneumothorax, pneumomediastinum, or bronchopulmonary fistula [12].
No guidelines exist for the management of IPPFE and there is mixed evidence supporting the use of steroids, immunosuppressants, and prophylactic antibiotics [4]. Further research is needed to identify effective treatment options for IPPFE. Several cases of successful lung transplantation have been reported [13, 14]. One review of 85 patients found a median survival rate of 11 years [15]. A rapidly progressive phenotype characterized by the coexistence of usual interstitial pneumonia (UIP) in the lower lobes has been described with a median survival rate of 24 months [16]. In this retrospective cohort study published by Kato et al in 2019, 36 patients with IPPFE between April 2009 and September 2017 were analyzed with an objective to determine the prognostic factors affecting the survival of patients with this rare disease [16]. Using HRCT scan, the multivariate analysis revealed that patients with UIP patterns in the lower lobes had a shorter survival time when compared to those with absence of this radiographic features. Unfortunately, our patient never had a HRCT done to help us determine if this was also going to be an independent prognostic factor that determined a timeline for her survival according to this study [16].
Our female patient with a BMI of 14.4 kg/m2 and a decreased anterior-posterior thoracic depth (188.3 mm) fits the classic demographic for IPPFE. She presented with the classic symptom of dyspnea and had a known history of recent unexplained pneumomediastinum. These features in conjunction with the patient’s classical upper lobe predominant interstitial lung disease and pleural thickening on CT scan enabled us to make the diagnosis of IPPFE. Given our patient’s frail body habitus, history of pneumomediastinum, and advanced age, the decision was made not to pursue surgical biopsy. Our patient did not respond to corticosteroid treatment, and she was not a candidate for a lung transplant. The rapidly progressive disease course seen in our patient supports the finding that female patients with low BMI are predisposed to developing a more severe phenotype of the disease.
Learning points
IPPFE is a devastating disease that may be more common than what is reported in the literature as a result of overlap with other interstitial lung disorders leading to misdiagnosis and unreliability in detection. More commonly seen in females with low BMI, as seen in our patient, median survival is only about 24 months. A rapidly progressive phenotype evident with UIP findings noted using HRCT scan has been proposed to be a major prognostic factor for patients survival associated with IPPFE. Further research is warranted to establish better criteria for diagnosis, as well as effective therapeutic interventions.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Informed consent was obtained from the patient before the presentation of this manuscript.
Author Contributions
Each author has been individually involved in and has made substantial contributions to conceptions and designs, acquisition of data, analysis, interpretation of data, drafting, and editing the manuscript. KT and SD contributed to the designs, acquisition of data, analysis, interpretation of data, drafting, and editing of the manuscript; MO contributed to the drafting and interpretation of data; JI and NU contributed to the drafting and editing of the manuscript; TK contributed to the designs, analysis, and editing of the manuscript; NS contributed to the analysis and editing of the manuscript; SC and AA contributed to the designs, analysis, interpretation of data, and editing of the manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Amitani R, Niimi A, Kuse F. Idiopathic pulmonary upper lobe fibrosis (IPUF). Kokyu. 1992;11:693-639.
- Travis WD, Costabel U, Hansell DM, King TE, Jr., Lynch DA, Nicholson AG, Ryerson CJ, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733-748.
doi pubmed - Reddy TL, Tominaga M, Hansell DM, von der Thusen J, Rassl D, Parfrey H, Guy S, et al. Pleuroparenchymal fibroelastosis: a spectrum of histopathological and imaging phenotypes. Eur Respir J. 2012;40(2):377-385.
doi pubmed - Chua F, Desai SR, Nicholson AG, Devaraj A, Renzoni E, Rice A, Wells AU. Pleuroparenchymal fibroelastosis. a review of clinical, radiological, and pathological characteristics. Ann Am Thorac Soc. 2019;16(11):1351-1359.
doi pubmed - Pakhale SS, Hadjiliadis D, Howell DN, Palmer SM, Gutierrez C, Waddell TK, Chaparro C, et al. Upper lobe fibrosis: a novel manifestation of chronic allograft dysfunction in lung transplantation. J Heart Lung Transplant. 2005;24(9):1260-1268.
doi pubmed - von der Thusen JH, Hansell DM, Tominaga M, Veys PA, Ashworth MT, Owens CM, Nicholson AG. Pleuroparenchymal fibroelastosis in patients with pulmonary disease secondary to bone marrow transplantation. Mod Pathol. 2011;24(12):1633-1639.
doi pubmed - Hirota T, Yoshida Y, Kitasato Y, Yoshimi M, Koga T, Tsuruta N, Minami M, et al. Histological evolution of pleuroparenchymal fibroelastosis. Histopathology. 2015;66(4):545-554.
doi pubmed - Nunes H, Jeny F, Bouvry D, Picard C, Bernaudin JF, Menard C, Brillet PY, et al. Pleuroparenchymal fibroelastosis associated with telomerase reverse transcriptase mutations. Eur Respir J. 2017;49(5):1602022.
doi pubmed - Shioya M, Otsuka M, Yamada G, Umeda Y, Ikeda K, Nishikiori H, Kuronuma K, et al. Poorer prognosis of idiopathic pleuroparenchymal fibroelastosis compared with idiopathic pulmonary fibrosis in advanced stage. Can Respir J. 2018;2018:6043053.
doi pubmed - Cuppens K, Verbeken E, Coolen J, Verschakelen J, Wuyts W. Idiopathic pleuroparenchymatous fibroelastosis: A case report and brief review of the literature. Respir Med Case Rep. 2014;12:7-9.
doi pubmed - Pickard A, Darby M, Soar J. Radiological assessment of the adult chest: implications for chest compressions. Resuscitation. 2006;71(3):387-390.
doi pubmed - Becker CD, Gil J, Padilla ML. Idiopathic pleuroparenchymal fibroelastosis: an unrecognized or misdiagnosed entity? Mod Pathol. 2008;21(6):784-787.
doi pubmed - Righi I, Morlacchi L, Rossetti V, Mendogni P, Palleschi A, Tosi D, Pieropan S, et al. Lung transplantation as successful treatment of end-stage idiopathic pleuroparenchymal fibroelastosis: a case report. Transplant Proc. 2019;51(1):235-238.
doi pubmed - Chen F, Matsubara K, Miyagawa-Hayashino A, Tada K, Handa T, Yamada T, Sato M, et al. Lung transplantation for pleuroparenchymal fibroelastosis after chemotherapy. Ann Thorac Surg. 2014;98(5):e115-117.
doi pubmed - Watanabe K. Pleuroparenchymal Fibroelastosis: Its Clinical Characteristics. Curr Respir Med Rev. 2013;9:299-237.
doi pubmed - Kato M, Sasaki S, Kurokawa K, Nakamura T, Yamada T, Sasano H, Arano N, et al. Usual interstitial pneumonia pattern in the lower lung lobes as a prognostic factor in idiopathic pleuroparenchymal fibroelastosis. Respiration. 2019;97(4):319-328.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.