

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 13, Number 6, June 2022, pages 290-296

A Continuous Increase in CXC-Motif Chemokine Ligand 10 in a Case of Anti-Nuclear Matrix Protein-2-Positive Juvenile Dermatomyositis
Tsunehisa Nagamoria, e, Emi Ishibazawaa, Yoichiro Yoshidaa, Kengo Izumib, Masayuki Satob, Yuki Ichimurac, Naoko Okiyamac, Ichizo Nishinod, Hiroshi Azumaa
aDepartment of Pediatrics, Asahikawa Medical University, Asahikawa, Hokkaido, Japan
bDepartment of Pediatrics, Asahikawa Kosei General Hospital, Asahikawa, Hokkaido, Japan
cDepartment of Dermatology, Faculty of Medicine, University of Tsukuba, Tsukuba, Ibaraki, Japan
dDepartment of Neuromuscular Research, National Institute of Neuroscience, National Center of Neurology and Psychiatry, Tokyo, Japan
eCorresponding Author: Tsunehisa Nagamori, Department of Pediatrics, Asahikawa Medical University, Asahikawa City, Hokkaido, Japan
Manuscript submitted April 8, 2022, accepted May 21, 2022, published online June 2, 2022
Short title: Intervention in Anti-NXP2-Positive JDM
doi: https://doi.org/10.14740/jmc3940
| Abstract | ▴Top |
Anti-nuclear matrix protein-2 (NXP2) antibody is associated with the severe, chronic myositis phenotype in juvenile dermatomyositis (JDM). Although hyperproduction of type I interferon is considered to play an important role in JDM, sequential changes in biomarkers associated with this pathophysiology have not yet been described in detail. An 8-year-old boy who presented with muscle weakness, heliotrope rash, and Gottron’s papules was diagnosed with JDM. With regard to myositis-specific autoantibodies, anti-NXP2 was detected. Although the increase of serum myogenic enzymes was modest at onset, two courses of methyl-prednisolone (mPSL) pulse therapy followed by oral prednisolone and methotrexate were insufficient to initiate remission. Therefore, additional treatment, with intravenous cyclophosphamide (IVCY) and intravenous immunoglobulin (IVIG) was required to obtain a favorable outcome. We also retrospectively evaluated serum concentration of several cytokines: interleukin (IL)-6, soluble tumor necrotizing factor receptor (sTNFR)-1, sTNFR-2, IL-18, and CXC-motif chemokine ligand (CXCL)-10. The cytokine profile of this patient at onset showed a CXCL-10-dominant pattern. Additionally, sequential evaluation of CXCL-10 revealed an aberrantly high level of CXCL-10 persistent despite two courses of mPSL pulse therapy, and the level of this cytokine only gradually decreased after initiation of IVCY and IVIG. The hyperproduction of CXCL-10, presumably reflecting the hyperproduction of type I interferon in the affected tissue, may persist for a certain period, even after the initiation of multiple courses of mPSL pulse therapy. With regard to the fact that anti-NXP2 is associated with subcutaneous calcification, our data suggest the importance of aggressive intervention in cases of anti-NXP2-positive JDM as well as the need for the development of a more pathophysiologically specific treatment.
Keywords: Juvenile dermatomyositis; Anti-nuclear matrix protein-2 antibody; CXC-motif chemokine ligand 10; Type I interferon
| Introduction | ▴Top |
Juvenile dermatomyositis (JDM) is a rare, systemic autoimmune disease characterized by inflammation, weakness of the proximal muscles, pathognomonic skin rashes, including heliotrope rash, and Gottron’s papules with onset during childhood [1]. Recent identification of novel myositis-specific autoantibodies (MSAs) frequently detected in JDM, such as anti-transcriptional intermediary factor 1γ (TIF1γ), anti-melanoma differentiation associated gene-5 (MDA5), and nuclear matrix protein-2 (NXP2), has provided further understanding of the clinical characteristics of JDM [2-4]. Anti-NXP2 antibody is detected in 15-23% of patients with JDM and is associated with a severe phenotype that involves contracture and weakness of the muscles and subcutaneous calcification [2, 3].
The pathophysiology of JDM is regarded as a combination of various mechanisms: vasculopathy based on autoimmunity and subsequent ischemic changes, activation of dendritic cells and upregulation of type I interferon, as well as endoplasmic reticulum stress in affected tissues [1]. Hyperproduction of type I interferon is regarded as the central pathogenesis of JDM, and can be applied to the assessment of disease activity and represents a potential target of future specific treatments for JDM. Here, we describe a case of anti-NXP2-positive JDM in which the patient presented with typical symptoms and was treated by glucocorticoids, methotrexate and additional intravenous cyclophosphamide (IVCY) and high-dose immunoglobulin. We demonstrate his clinical course, histological findings, as well as serum cytokine profile and the time course of changes in CXC-motif chemokine ligand (CXCL)-10, an interferon-induced protein.
| Case Report | ▴Top |
Investigations
An 8-year-old boy presented with a 3-month history of muscle weakness and a rash on his limbs and eyelids. He had progressive muscle weakness which left him unable to get up from a squatting position without support of his hands, and he was also unable to open the screw cap on plastic bottles. On examination, he had low-grade fever, weakness of the proximal muscles of the legs and arms, heliotrope rash, Gottron’s papules and erythema over his knees and elbows (Fig. 1a-c). His height was 118 cm (-1.2 SD) and body weight was 19.4 kg, giving a body mass index of 13.8. Laboratory findings (Table 1) showed elevated aspartate aminotransferase (AST), lactate dehydrogenase (LDH), and aldolase levels, although the creatine phosphokinase (CPK) level was within the normal range. Although the patient was negative for C-reactive protein, his erythrocyte sedimentation rate (ESR) and ferritin, serum β2-microglobulin (β2MG), and soluble interleukin-2 receptor (sIL-2R) levels were elevated. The antinuclear antibody (ANA) titer was 1:640, and he was positive for the anti-NXP2 autoantibody evaluated through immunoprecipitation and Western blotting [5], but negative for other autoantibodies such anti-SS-A, anti-SS-B, anti-Sm, anti-ribonucleoprotein (RNP), anti-double-stranded DNA, anti-Jo-1, anti-Mi-2, anti-MDA5, and anti-TIF1γ. The patient underwent magnetic resonance imaging for high-intensity areas throughout all skeletal muscles using T2-weighted and short tau inversion recovery (STIR) images (Fig. 1d). The childhood myositis assessment scale (CMAS) [6] was scored as 23, although there was difficulty in obtaining sufficient cooperation.
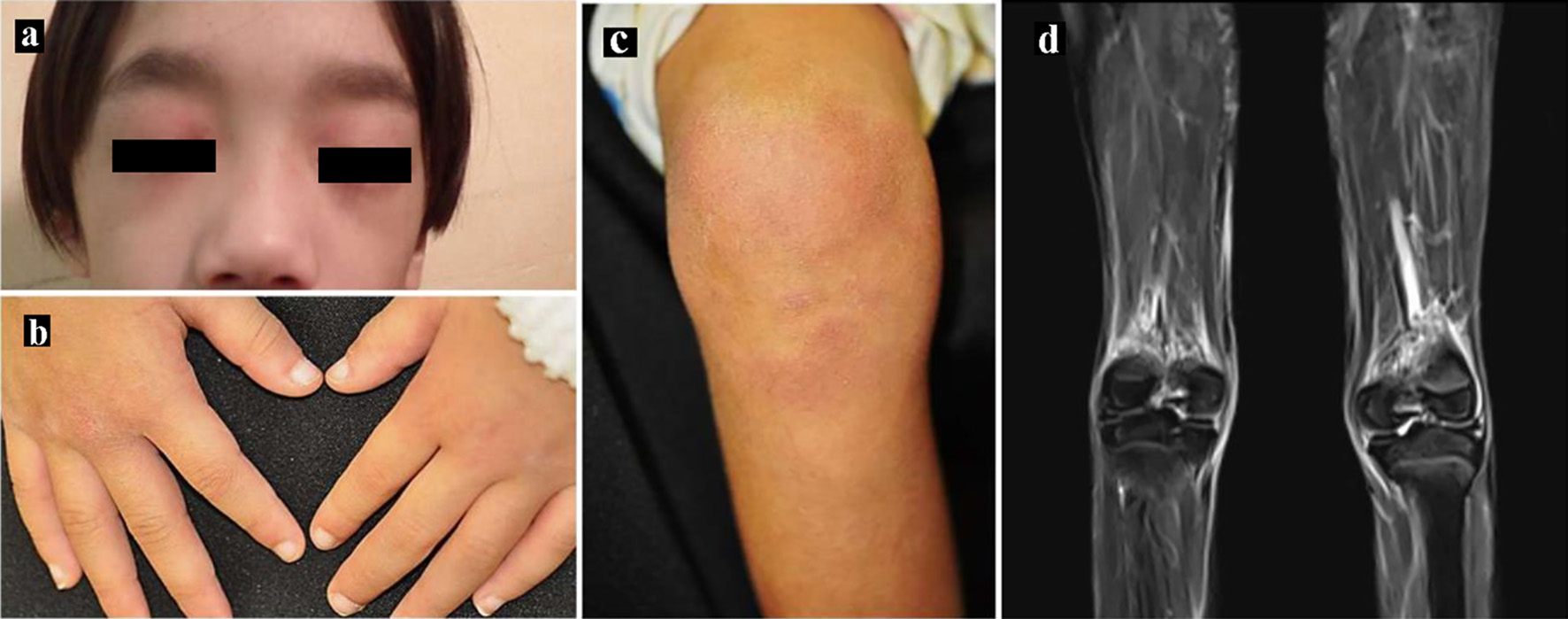 Click for large image | Figure 1. Skin manifestations. (a) Heliotrope rash, (b) Gottron’s papules and (c) erythema over the knees. (d) Magnetic resonance imaging showing T2-weighted and short tau inversion recovery (STIR) images of the lower limbs. |
 Click to view | Table 1. Laboratory Results |
We carried out muscle biopsy from the left biceps brachii muscle, with the histological findings as follows. Hematoxylin and eosin staining revealed a collection of atrophic fibers with basophilic cytoplasm and enlarged nuclei in the perifascicular areas, indicating perifascicular atrophy. No apparent endomysial fibrosis was observed. Minimal lymphocyte infiltration was observed in the perivascular area, and macrophages were scattered throughout the perimysium (Fig. 2a). Immunohistochemical analysis revealed, in addition to the expression of human leukocyte antigen (HLA)-ABC with perifascicular enhancement, the expression of myxovirus-resistance protein A (MxA), which is a diagnostic marker of dermatomyositis in terms of muscle pathology [7], and membrane attack complex (MAC) deposition in the endomysial capillaries, which is also diagnostic of dermatomyositis (Fig. 2b-d).
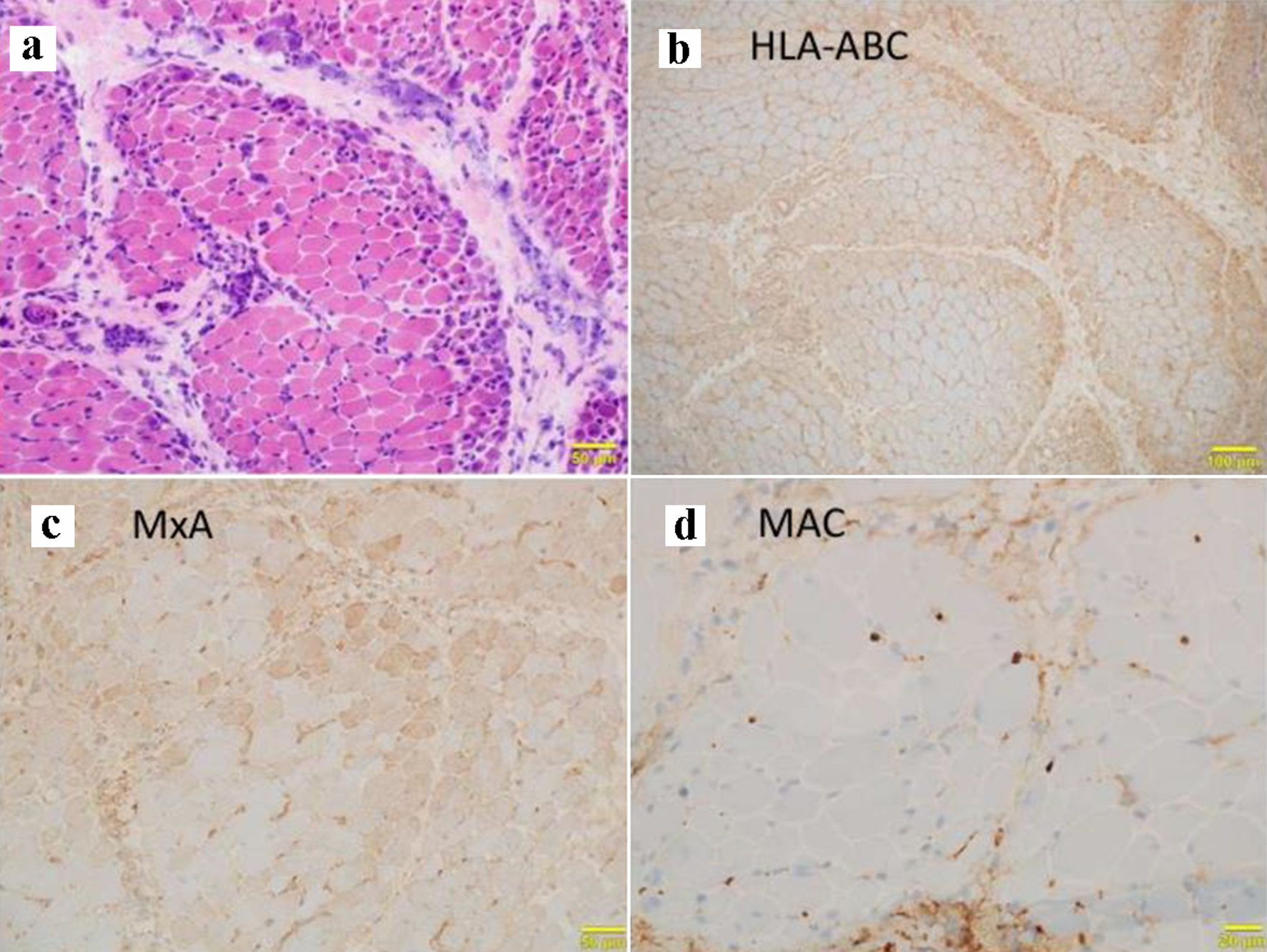 Click for large image | Figure 2. Muscle biopsy findings. (a) Hematoxylin and eosin staining showing perifascicular atrophy with atrophic basophilic fibers at the periphery of the fascicles in addition to minimal perivascular infiltration of lymphocytes. Immunohistochemical staining for (b) human leukocyte antigen (HLA)-ABC, (c) myxovirus-resistance protein A (MxA) showing the expression of interferon-induced protein, and (d) membrane attack complex (MAC) showing complement deposition in the endomysial capillaries. |
Diagnosis
Based on the observations of typical skin rush and muscle weakness, increase in serum myogenic enzyme, positivity for anti-NXP2 antibody, and typical histological findings, we diagnosed the patient with JDM.
Treatment
We then administered two courses of methyl-prednisolone (mPSL) pulse therapy followed by 1 mg/kg/day prednisolone and weekly oral methotrexate (MTX). Laboratory findings of muscle-derived enzymes showed transient improvement after treatment; however, a subsequent increase in aldolase was observed. We, therefore, additionally provided six courses of IVCY and intravenous immunoglobulin (IVIG) therapy. As a result, the laboratory findings for aldolase, β2MG, and sIL-2R gradually improved and oral prednisolone was successfully decreased.
Follow-up and outcomes
The patient’s muscle weakness also gradually improved, and his CMAS was about 37 at 4 months and 41 at 9 months after admission, which was sufficient for activities of daily life. He has been followed up for 24 months to date, and although one mild relapse occurred, presumably due to non-adherence to the medication, the patient has been stable and has not developed calcinosis to date.
Additional investigations
We also retrospectively evaluated the serum concentration of several cytokines using the Bioplex multiplex assay system (Bio-RAD, CA, USA). Human cytokine assay kits for IL-6 (#171B5006M), soluble tumor necrotizing factor receptor (sTNFR)-1, sTNFR2 (#171BL034M and #171BL035M), IL-18 (#171B6023M) and CXCL-10 (#171B5020M) were individually purchased and each cytokine was mixed and evaluated in accordance with the manufacturer’s recommendations. The assays were performed on serum samples from healthy adults (n = 8), from a patient with systemic juvenile idiopathic arthritis (sJIA) (n = 1, four samples obtained before treatment initiation), patients with Kawasaki disease (n = 6), and this patient (two samples obtained before treatment initiation, and a further 16 consecutive samples). Compared to healthy adults, the cytokine profile of the sJIA showed an IL-18-dominant pattern, while that for Kawasaki disease showed an increase in IL-6 and sTNFR, which was consistent with previously reported results [8, 9] (Fig. 3a). The cytokine profile for our patient before treatment initiation showed a noticeably CXCL-10-dominant pattern (Fig. 3a). Additionally, the time course of CXCL-10 concentration showed continuing elevation. Interestingly, although transient decreases in serum CXCL-10 after the two courses of mPSL pulse were observed, it remained increased for a certain period, and only gradually decreased after the administration of IVCY and IVIG (Fig. 3b).
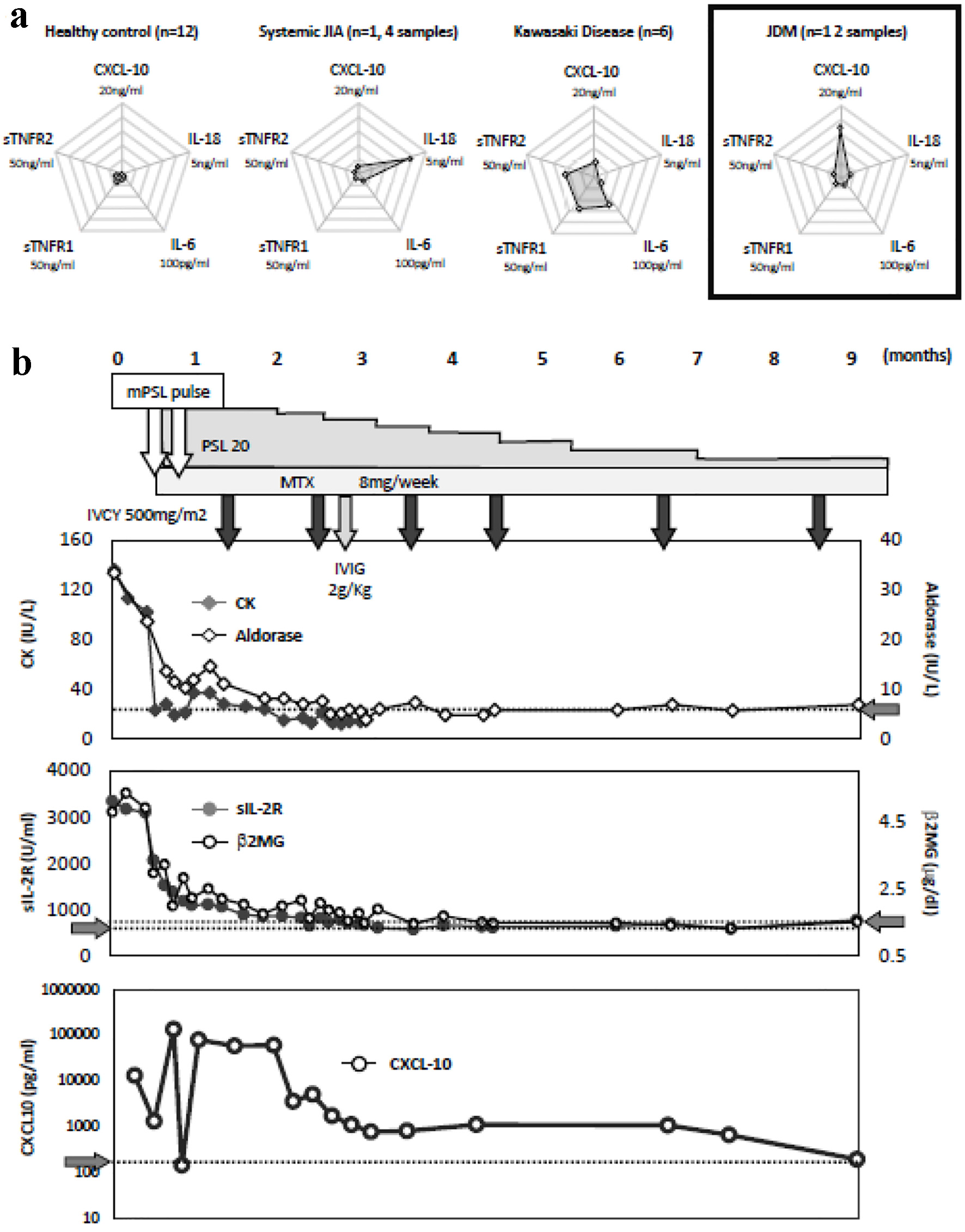 Click for large image | Figure 3. (a) Cytokine profiling of healthy adults (n = 8), a patient with systemic juvenile idiopathic arthritis (sJIA) (n = 1, four samples obtained before treatment initiation), patients with Kawasaki disease (n = 6), and this patient (two samples obtained before treatment initiation, and a further 16 consecutive samples). (b) Clinical course of the patient. Content of treatment, sequential changes in serum creatine kinase (CK, IU/L), aldolase (IU/L), soluble interleukin-2 receptor (sIL-2R, IU/L) beta-2 microglobulin (β2MG, µg/dL) levels, as well as retrospectively evaluated serum CXC-motif chemokine ligand (CXCL)-10 levels (pg/mL) are shown. The gray arrows on the vertical axis indicate the upper limits of Aldorase, sIL-2R, and β2MG, as well as the average CXCL-10 in healthy adults. |
| Discussion | ▴Top |
JDM is a heterogeneous disease and autoantibodies are potentially useful biomarkers by which to divide patients into homogeneous subgroups and provide information on prognosis. Anti-NXP2 was first identified from a JDM cohort and reported in 1997 as anti-MJ. Espada et al [10] suggested an association between anti-NXP2 and a significant compromise in functional status characterized by muscle contracture and atrophy. Furthermore, studies of other independent cohorts in France and the UK subsequently revealed that anti-NXP2 was associated with a severe subtype of JDM in which patients developed severe muscular disease with a lower remission rate [3, 11]. Furthermore, the association between anti-NXP2 and a high frequency of calcinosis was reported [3, 12]. Additionally, several risk factors for a refractory clinical course among patients with anti-NXP2-positive JDM have been reported. Wang et al [13] reported an analysis of 26 anti-NXP2-positive JDM cases and found that edema, skin ulcers, significant muscle weakness, low CD4/CD8 ratio and high serum ferritin level at disease onset were associated with a refractory clinical course. Importantly, the serum levels of muscular enzymes at onset did not provide a basis for differentiating clinical outcomes. In our case, the increases in serum muscular enzymes were only modest, and no edema or ulcers were observed. On the other hand, significant muscle weakness and increases in serum ferritin level were observed, and the response to initial treatment was limited. It appeared that our patient was at a comparatively high risk and aggressive treatment including IYCY, IVIG in addition to mPSL pulse and short MTX therapy was necessary.
The association of type I interferon with JDM pathophysiology has been the subject of a considerable amount of researches. Plasmacytoid dendritic cells were shown to be the predominant subsets of inflamed muscle tissue [14], and serum interferon-α activity was also reported to be elevated [15]. The global gene expression pattern in the peripheral blood cells of JDM patients shows a subset of type I interferon-inducible genes [16, 17]. To investigate the pathophysiology, we carried out cytokine assays of IL-6, sTNFR1, sTNFR2, IL-18 and CXCL-10 [18], and observed a solely CXCL-10-dominant pattern. As CXCL-10 is a chemokine that attracts lymphocytes to production sites and, at the same time, belongs to the interferon-induced protein family, the increase in CXCL-10 may be the result of excessive type I interferon. In fact, a previous report showed an increase in serum CXCL-10 along with disease activity in JDM patients [19]. Additionally, we found that the increase in serum CXCL-10 was more prolonged than expected, and persisted even after multiple courses of mPSL pulse therapy. This indicated that the disease activity of JDM lasted for a certain period, even after the serum concentrations of myogenic enzymes were within their normal ranges.
The development of calcinosis in JDM is associated with delayed diagnosis, a chronic disease course and inadequately treated disease [20, 21]. It is also associated with a lower age at disease onset and anti-NXP2 positivity [3, 12]. As calcification is regarded to result from chronic inflammation, our finding showing a persistent increase in CXCL-10 in a patient with JDM with anti-NXP2 antibodies who received comparatively aggressive treatment might have an impact on the future management of this disease in terms of the importance of the adequate induction of clinical remission. Further, our findings appear to support the further progress toward to more disease-specific agents to reduce the interferon-induced pathophysiology, such as Janus-kinase inhibitors, which have recently been reported to be effective in the treatment of JDM [22, 23]. Further investigation is required to prove this hypothesis.
Learning points
A continuous increase in CXCL-10 in this patient implies the need for aggressive intervention in anti-NXP2-positive JDM patients. Moreover, the development of a pathophysiologically specific treatment is desirable.
Acknowledgments
None to declare.
Financial Disclosure
This study was supported partly by Intramural Research Grant (2-5) for Neurological and Psychiatric Disorders from the National Center of Neurology and Psychiatry, and JSPS KAKENHI Grant Number 21K07836.
Conflict of Interest
None to declare.
Informed Consent
We obtained written informed consent for the use of clinical data and the preserved serum samples from the parents of the patient.
Author Contributions
TN, EI, YY, KI, and MS obtained clinical data. TN, EI, and YY carried out the evaluation of cytokine profiling. TN and EI wrote the draft of the main manuscript. IN provided histological evaluations. YI and NO evaluated anti-NXP2 through immunoprecipitation and Western blotting. HA gave instructions throughout the entire procedure.
Data Availability
The authors declare that the data supporting this study are available within the article.
| References | ▴Top |
- Feldman BM, Rider LG, Reed AM, Pachman LM. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet. 2008;371(9631):2201-2212.
doi - Alenzi FM. Myositis Specific Autoantibodies: A Clinical Perspective. Open Access Rheumatol. 2020;12:9-14.
doi pubmed - Tansley SL, Betteridge ZE, Shaddick G, Gunawardena H, Arnold K, Wedderburn LR, McHugh NJ, et al. Calcinosis in juvenile dermatomyositis is influenced by both anti-NXP2 autoantibody status and age at disease onset. Rheumatology (Oxford). 2014;53(12):2204-2208.
doi pubmed - Nakashima R. Clinical significance of myositis-specific autoantibodies. Immunol Med. 2018;41(3):103-112.
doi pubmed - Ichimura Y, Matsushita T, Hamaguchi Y, Kaji K, Hasegawa M, Tanino Y, Inokoshi Y, et al. Anti-NXP2 autoantibodies in adult patients with idiopathic inflammatory myopathies: possible association with malignancy. Ann Rheum Dis. 2012;71(5):710-713.
doi pubmed - Lovell DJ, Lindsley CB, Rennebohm RM, Ballinger SH, Bowyer SL, Giannini EH, Hicks JE, et al. Development of validated disease activity and damage indices for the juvenile idiopathic inflammatory myopathies. II. The Childhood Myositis Assessment Scale (CMAS): a quantitative tool for the evaluation of muscle function. The Juvenile Dermatomyositis Disease Activity Collaborative Study Group. Arthritis Rheum. 1999;42(10):2213-2219.
doi - Uruha A, Nishikawa A, Tsuburaya RS, Hamanaka K, Kuwana M, Watanabe Y, Suzuki S, et al. Sarcoplasmic MxA expression: A valuable marker of dermatomyositis. Neurology. 2017;88(5):493-500.
doi pubmed - Jinkawa A, Shimizu M, Nishida K, Kaneko S, Usami M, Sakumura N, Irabu H, et al. Cytokine profile of macrophage activation syndrome associated with Kawasaki disease. Cytokine. 2019;119:52-56.
doi pubmed - Shimizu M, Yokoyama T, Yamada K, Kaneda H, Wada H, Wada T, Toma T, et al. Distinct cytokine profiles of systemic-onset juvenile idiopathic arthritis-associated macrophage activation syndrome with particular emphasis on the role of interleukin-18 in its pathogenesis. Rheumatology (Oxford). 2010;49(9):1645-1653.
doi pubmed - Espada G, Maldonado Cocco JA, Fertig N, Oddis CV. Clinical and serologic characterization of an Argentine pediatric myositis cohort: identification of a novel autoantibody (anti-MJ) to a 142-kDa protein. J Rheumatol. 2009;36(11):2547-2551.
doi pubmed - Aouizerate J, De Antonio M, Bader-Meunier B, Barnerias C, Bodemer C, Isapof A, Quartier P, et al. Muscle ischaemia associated with NXP2 autoantibodies: a severe subtype of juvenile dermatomyositis. Rheumatology (Oxford). 2018;57(5):873-879.
doi pubmed - Gunawardena H, Wedderburn LR, Chinoy H, Betteridge ZE, North J, Ollier WE, Cooper RG, et al. Autoantibodies to a 140-kd protein in juvenile dermatomyositis are associated with calcinosis. Arthritis Rheum. 2009;60(6):1807-1814.
doi pubmed - Wang X, Ding Y, Zhou Z, Hou J, Xu Y, Li J. Clinical characteristics and poor predictors of anti-NXP2 antibody-associated Chinese JDM children. Pediatr Rheumatol Online J. 2021;19(1):6.
doi pubmed - Lopez de Padilla CM, Vallejo AN, McNallan KT, Vehe R, Smith SA, Dietz AB, Vuk-Pavlovic S, et al. Plasmacytoid dendritic cells in inflamed muscle of patients with juvenile dermatomyositis. Arthritis Rheum. 2007;56(5):1658-1668.
doi pubmed - Niewold TB, Kariuki SN, Morgan GA, Shrestha S, Pachman LM. Elevated serum interferon-alpha activity in juvenile dermatomyositis: associations with disease activity at diagnosis and after thirty-six months of therapy. Arthritis Rheum. 2009;60(6):1815-1824.
doi pubmed - Baechler EC, Bilgic H, Reed AM. Type I interferon pathway in adult and juvenile dermatomyositis. Arthritis Res Ther. 2011;13(6):249.
doi pubmed - Rider LG, Nistala K. The juvenile idiopathic inflammatory myopathies: pathogenesis, clinical and autoantibody phenotypes, and outcomes. J Intern Med. 2016;280(1):24-38.
doi pubmed - Nagamori T, Yoshida Y, Ishibazawa E, Oka H, Takahashi H, Manabe H, Taketazu G, et al. Variations in the pathophysiology of respiratory syncytial virus infection depend on the age at onset. Pediatr Int. 2022;64(1):e14720.
doi pubmed - Bellutti Enders F, van Wijk F, Scholman R, Hofer M, Prakken BJ, van Royen-Kerkhof A, de Jager W. Correlation of CXCL10, tumor necrosis factor receptor type II, and galectin 9 with disease activity in juvenile dermatomyositis. Arthritis Rheumatol. 2014;66(8):2281-2289.
doi pubmed - Marhaug G, Shah V, Shroff R, Varsani H, Wedderburn LR, Pilkington CA, Brogan PA. Age-dependent inhibition of ectopic calcification: a possible role for fetuin-A and osteopontin in patients with juvenile dermatomyositis with calcinosis. Rheumatology (Oxford). 2008;47(7):1031-1037.
doi pubmed - Hoeltzel MF, Oberle EJ, Robinson AB, Agarwal A, Rider LG. The presentation, assessment, pathogenesis, and treatment of calcinosis in juvenile dermatomyositis. Curr Rheumatol Rep. 2014;16(12):467.
doi pubmed - Le Voyer T, Gitiaux C, Authier FJ, Bodemer C, Melki I, Quartier P, Aeschlimann F, et al. JAK inhibitors are effective in a subset of patients with juvenile dermatomyositis: a monocentric retrospective study. Rheumatology (Oxford). 2021;60(12):5801-5808.
doi pubmed - Yu Z, Wang L, Quan M, Zhang T, Song H. Successful management with Janus kinase inhibitor tofacitinib in refractory juvenile dermatomyositis: a pilot study and literature review. Rheumatology (Oxford). 2021;60(4):1700-1707.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.