

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 14, Number 5, May 2023, pages 162-168

Vanishing Bile Duct Syndrome in a Patient With Recurrent Hodgkin Lymphoma
Olger Nanoa, b, d, Stanislav Ivanova, b, Tarun Kapoorc
aDepartment of Hematology and Oncology, Memorial Healthcare System, Hollywood, FL 33021, USA
bDepartment of Hematology and Oncology, Memorial Cancer Institute, Pembroke Pines, FL 33026, USA
cWake Forest University School of Medicine, Winston-Salem, NC 27101, USA
dCorresponding Author: Olger Nano, Department of Hematology and Oncology, Memorial Cancer Institute, Pembroke Pines, FL 33026, USA
Manuscript submitted March 17, 2023, accepted May 6, 2023, published online May 31, 2023
Short title: VBDS With Recurrent Hodgkin Lymphoma
doi: https://doi.org/10.14740/jmc4073
| Abstract | ▴Top |
Vanishing bile duct syndrome (VBDS) is an acquired syndrome characterized by clinical and laboratory signs of cholestasis with pathologic findings of interlobular bile duct paucity in liver biopsy specimens. VBDS can result from a variety of conditions including infections, autoimmune diseases, adverse drug reactions, and neoplastic processes. Hodgkin lymphoma (HL) is a rare cause of VBDS. The mechanism by which HL leads to VBDS remains unknown. Development of VBDS in patients with HL portends an extremely poor prognosis due to the risk of progression to fulminant hepatic failure. Treatment of the underlying lymphoma has been demonstrated to offer increased probability of recovery from VBDS. The decision to treat and choice of treatment of the underlying lymphoma is often complicated by the hepatic dysfunction characteristic of VBDS. We present the case of a patient who presented with dyspnea and jaundice in the context of recurrent HL and VBDS. We additionally review the literature on HL complicated by VBDS with specific focus on treatment paradigms for management of these patients.
Keywords: Vanishing bile duct syndrome; VBDL; Hodgkin lymphoma; Hepatic failure
| Introduction | ▴Top |
Vanishing bile duct syndrome (VBDS) is an acquired syndrome which can be seen as a result of numerous underlying causes including genetic diseases (cystic fibrosis, trisomy 17, 18, 21 and Alagille syndrome), infectious etiologies (human immunodeficiency virus, cytomegalovirus, Epstein-Barr virus, and cryptosporidium infections), autoimmune conditions (sarcoidosis, primary biliary cirrhosis, primary sclerosing cholangitis (PSC), graft-versus-host disease, acute/chronic liver transplant rejection), adverse drug reactions (including weight loss supplements, anti-epileptics, and antibiotics), and neoplastic processes (including both Hodgkin and non-Hodgkin lymphoma as well as histiocytosis X) [1]. VBDS is a clinicopathologic diagnosis characterized by clinical and laboratory signs of cholestasis (jaundice, conjugated bilirubinemia, elevated alkaline phosphatase (ALP) and gamma-glutamyltransferase (GGT)) and biopsy-proven paucity of intrahepatic bile ducts [1, 2]. VBDS is a diagnosis of exclusion, requiring a high index of suspicion and exclusion of other common causes of cholestasis as discussed previously.
Hodgkin lymphoma (HL) is a monoclonal lymphoid neoplasm that classically affects young adults with an incidence of 2.6 cases per 100,000 people in the United States [3]. HL commonly arises in cervical, mediastinal, or axillary lymph nodes, and classic biopsy findings show large mononuclear Reed-Sternberg cells intermixed with non-neoplastic inflammatory cells (most often T lymphocytes). Classically, HL patients present with bulky lymphadenopathy; however, patients can present without evidence of palpable adenopathy [3, 4]. Hepatic involvement with HL is relatively common with reported lymphomatous infiltration rates of 5-8% in liver biopsies and as high as 30-70% in autopsy studies in HL patients [5-10]. Presentation with cholestasis as the sole presenting symptom in HL is uncommon (less than 4%) and can be due to lymphomatous infiltration into the liver extrahepatic bile duct, compression from bulky abdominal/periportal lymphadenopathy, or rarely VBDS.
VBDS is an exceedingly unusual complication of HL with approximately 50 cases reported in the literature. HL as a cause of VBDS was first recognized by Hubscher et al in 1993 when they described three patients with severe intrahepatic cholestasis (IC) and liver biopsies consistent with VBDS [11]. Prior to this, similar presentations of patients with jaundice and cholestasis in patients with HL had been reported and were diagnosed as “intrahepatic cholestasis” secondary to HL. The mechanism by which HL leads to VBDS is still not known. Several theories have been proposed including microscopic infiltration of lymphoma cells into the liver, release of cytotoxic cytokines from lymphoma cells leading to direct and/or indirect (e.g., T cell-mediated) destruction of bile duct epithelial cells. The latter theory is favored as biopsy and autopsy specimens have noted absence of lymphoma cell infiltrating the livers of patients with VBDS and HL.
Treatment for management of classical HL is based on an anthracycline-based regimens of chemotherapy like adriamycin, bleomycin, vinblastine, and dacarbazine (ABVD). Careful consideration must be given to the choice of therapy used for the management of classical HL, as many of the mainstay agents used are heavily metabolized via the liver. As such, these agents are contraindicated in patients with hepatic dysfunction, including bilirubin > 5 mg/dL. [12, 13]. These considerations pose difficulty in treating patients with HL and VBDS. Management of hyperbilirubinemia with agents like ursodeoxycholic acid and cholestyramine, which has shown promise in other etiologies like primary biliary cirrhosis (PBC) and IC of pregnancy, has had mixed reports when it comes to the management of VBDS, posing yet another hurdle in the management of this condition [14-20].
| Case Report | ▴Top |
Investigations
A 40-year-old male patient presented to the emergency department with 1 week of worsening shortness of breath, cough, and chest pain. He complained of exertional chest pain and shortness of breath, but denied diaphoresis, palpitations, or headaches. His past medical history was remarkable for hypertension (HTN), alcohol abuse, and HL, which was initially diagnosed 4 years prior to this presentation and was treated with ABVD.
Vitals on admission were notable for temperature of 37.1 °C, blood pressure of 131/73 mm Hg, heart rate of 109 beats per minute, respiratory rate of 18 per minute with oxygen saturation of 99% at room air. Physical exam was remarkable for findings of jaundice, scleral icterus as well as decreased breath sounds over the left lower lobe.
Review of the patient’s record was notable for two inpatient admissions 2 months prior to the current presentation. During these admissions the patient was extensively worked up for evidence of recurrent HL. The patient underwent a mediastinoscopy and lymph node biopsy which were negative for evidence of disease. Flow cytometry from the specimens were inconclusive. The patient developed transaminitis with elevated alanine transaminase (ALT), aspartate aminotransferase (AST) and markedly elevated ALP. The patient underwent evaluation with magnetic resonance cholangiopancreatography (MRCP) which was negative for evidence of obstruction. Subsequent liver biopsy demonstrated no evidence of progressive disease involvement. The patient was lost to follow-up prior to completing outpatient follow-up with endoscopic retrograde cholangiopancreatography (ERCP).
The patient presented once again to the emergency room with complaint of abdominal pain and shortness of breath. Vital signs were stable. Physical examination was once again remarkable for jaundice and scleral icterus. Computed tomography (CT) scan of the abdomen was unremarkable when compared to previous scan obtained at the time of his original diagnosis of HL. The patient was noted to have peri-aortic and abdominal lymph nodes consistent with lymphoma (Figs. 1, 2) Complete blood count on admission were concerning for leukocytosis with elevated white blood count of 47,000/µL with 86% neutrophils and 2% bands, mild anemia with hemoglobin of 8.8 mg/dL, hematocrit of 26.8%, and thrombocytosis with platelet count of 425,000/µL. Comprehensive metabolic panel was significant for hyponatremia with serum sodium of 127, hypochloremia with chloride at 97, alkalemia with bicarbonate of 16. Serum creatinine could not be calculated initially given the severely icteric sample. His ALP significantly increased to 1,876 U/L from 1,045 U/L on last discharge, with relatively stable ALT and AST. His total bilirubin was also markedly elevated to 28.2 mg/dL, with 23.3 mg/dL classified as direct bilirubin. This demonstrated significant change from previous values noted during his last admission, 12.1mg/dL and 10.1mg/dL, respectively. ALP was also noted to be markedly elevated at 1,761 U/L.
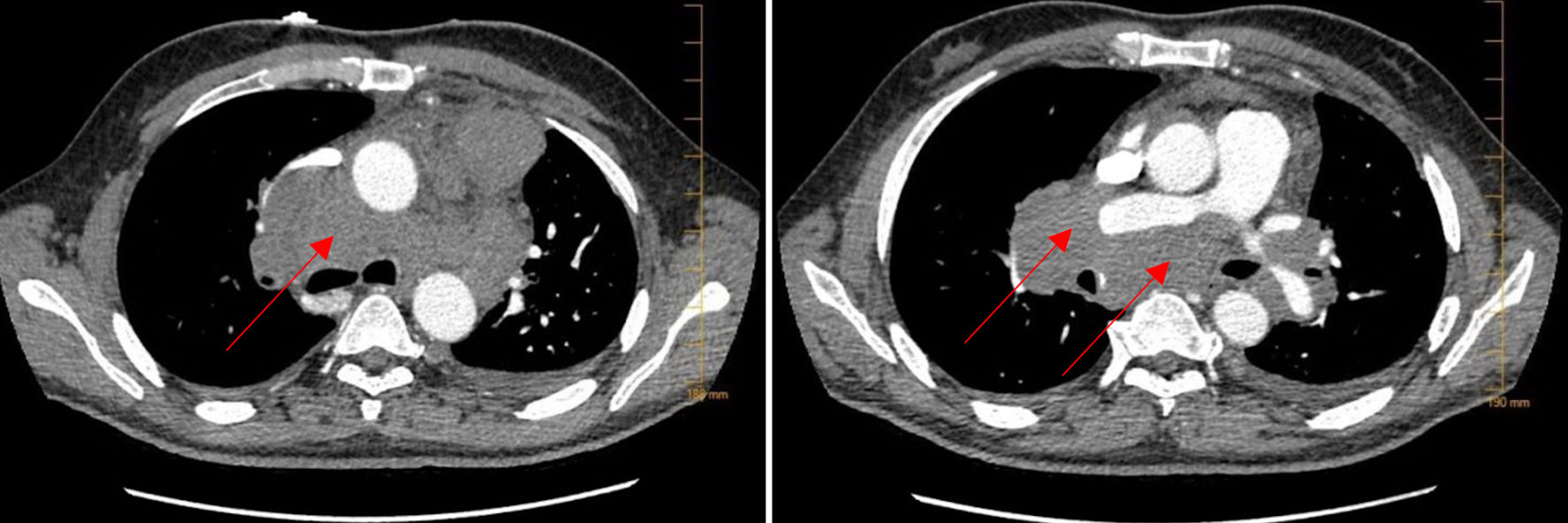 Click for large image | Figure 1. CTA of the lungs. CTA of the lungs on admission showed extensive bulky mediastinal and hilar lymphadenopathy with encasement of the pulmonary artery (arrows). CTA: computed tomography angiography. |
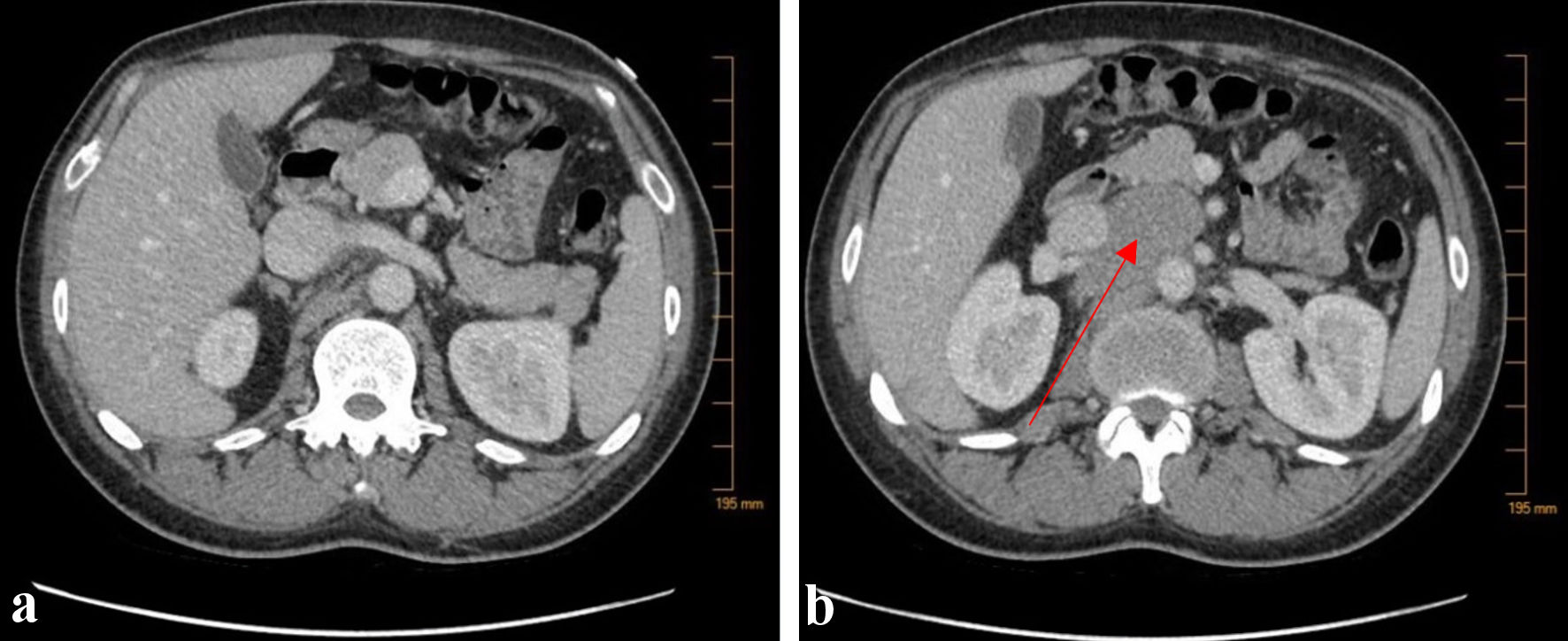 Click for large image | Figure 2. CT abdomen/pelvis with contrast. (a) CT abdomen/pelvis demonstrated a normal appearing liver without evidence of biliary tract dilation. (b) Additional bulky lymphadenopathy was also noted in the aortocaval node region as well as para-diaphragmatic nodes (arrow). CT: computed tomography. |
Repeat CT of the chest revealed interval development of bilateral pleural effusions along with lymphadenopathy and an increasing pericardial effusion (Fig. 3). Ultrasound-guided thoracentesis was performed yielding 1 L of yellow purulent fluid. His pleural fluid by Light’s criteria was consistent with exudative fluid (total protein was 5.8, lactate dehydrogenase (LDH) was 385 (range from our labs is 87 - 241), body fluid protein is 3.1, body fluid LDH was 141. Nevertheless, the fluid collected did not show any significant findings of malignant cells.
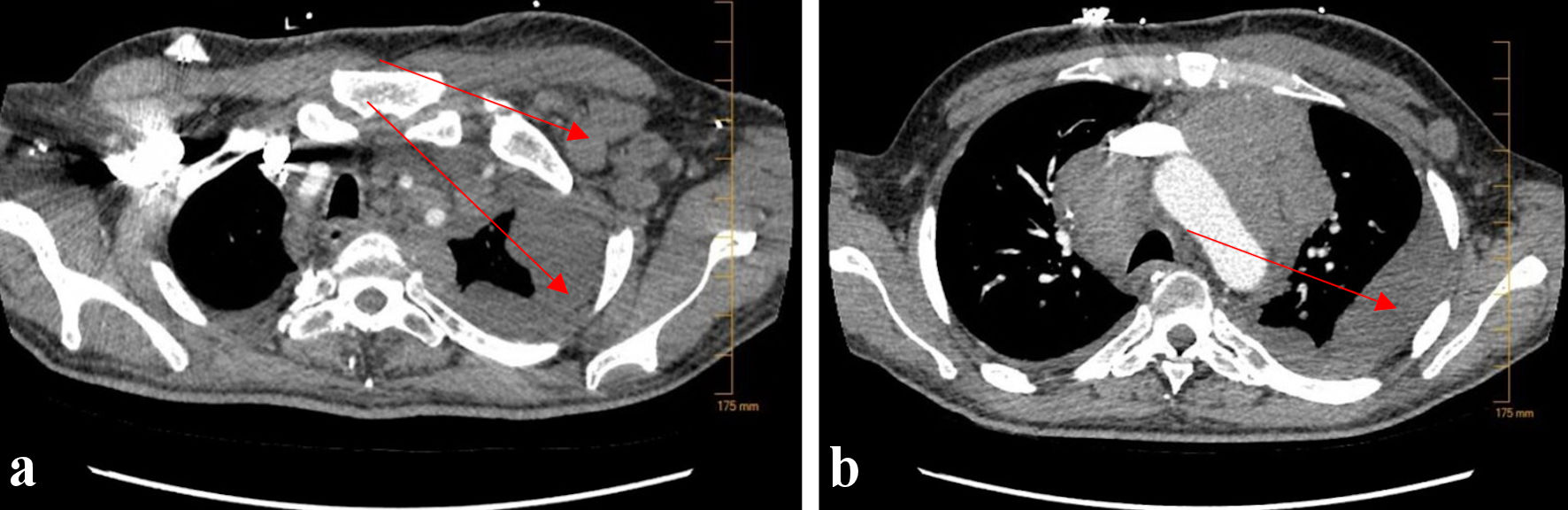 Click for large image | Figure 3. CTA of the lungs upon re-admission 2 months later. Repeat CTA of the lungs showed new, bulky left-sided axillary lymphadenopathy (a) (arrow), as well as extensive, worsening left-sided pleural effusion (a, b) (arrows). CTA: computed tomography angiography. |
A new biopsy was performed of the left axillary node which was compared to a biopsy of anterior tracheal mass obtained 4 years prior at the time of HL diagnosis. The results of the biopsy were once again noted to be positive for CD45, CD15, CD30, PAX-5, Fascin and negative for epithelial membrane antigen (EMA) and CD20, consistent with classic HL.
Immunohistology staining showed negative staining for pancytokeratin, synaptophysin, chromogranin, CD56, Mart 1, cytokeratin 7 and cytokeratin 20; but it was diffusely positive for leukocyte common antigen (LCA). Additional immunohistochemical stains were negative for CD15/CD30 with no evidence of Reed-Sternberg cells, PAX 5, CD20 (B cells), cyclin D1, CD23, CD10; it was positive for CD3 (T cells) and CD5. B cell receptor gamma gene rearrangements were also not detected, along with no detection of T cell receptor gamma gene rearrangements and T cell receptor beta gene rearrangements.
Pericardial fluid was not significant enough for pericardiocentesis. The patient’s renal function was impaired with creatinine increasing from 1.84 on admission (baseline of 0.6) up to 2.7. This was attributed to be secondary to bile acid nephropathy which was confirmed on urinalysis findings.
Diagnosis
To definitively establish a diagnosis, an ERCP was performed during this hospitalization showing sclerosing cholangitis and atrophy of the intrahepatic bile duct (Fig. 4). Mitochondrial antibody and immunoglobulin G4 (IgG4) were negative. The biopsy collected during ERCP showed portal tracts with mild chronic inflammation with no chronic fibrosis. No metaplastic bile ducts were seen. Of the 13 portal tracts evaluated, none contained significant fibrosis (Fig. 5). No plasma cell infiltrates were seen. Trichrome stain showed no definitive concentric fibrosis but did demonstrate sub-sinusoidal fibrosis. Microscopic findings were consistent with VBDS, which can be seen in patient with HL. Given the constellation of presenting symptoms as well as notable laboratory findings, a diagnosis of VBDS was made with etiology attributed to underlying HL.
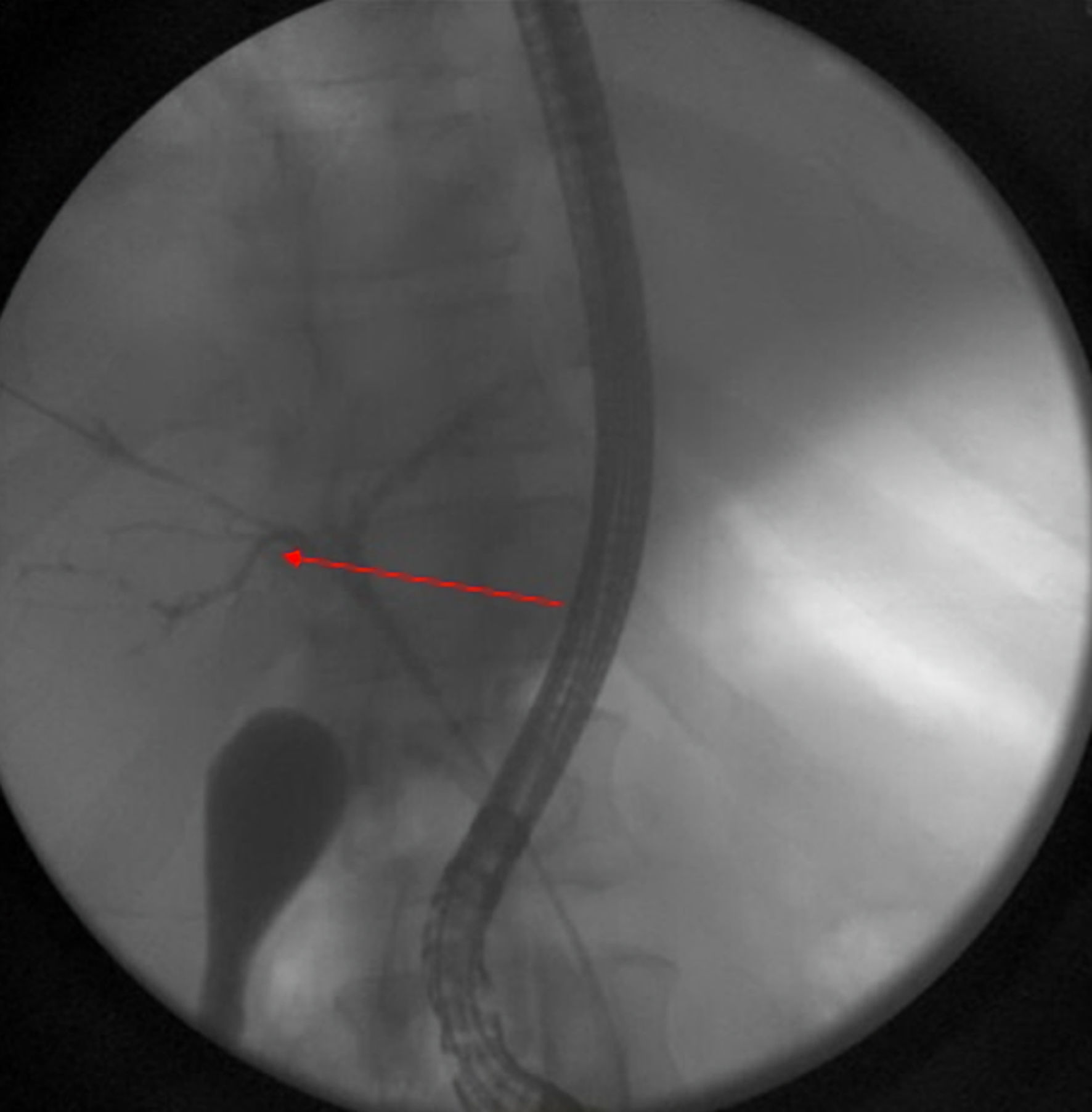 Click for large image | Figure 4. Still image from ERCP study demonstrating failure of contrast penetration through the biliary tree. Findings additionally demonstrate atrophy of intrahepatic ducts with appearance of sclerosing cholangitis (arrow). ERCP: endoscopic retrograde cholangiopancreatography. |
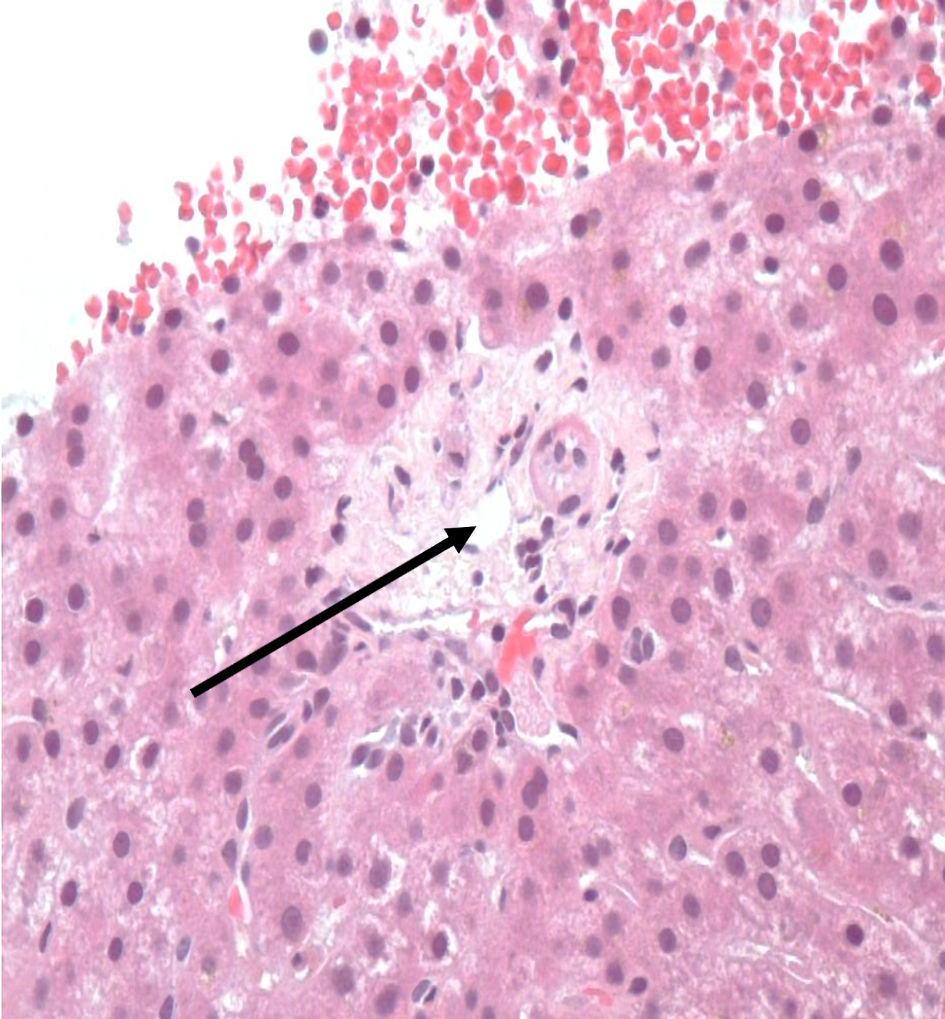 Click for large image | Figure 5. Liver biopsy demonstrating absence of interlobular bile duct and hepatocyte injury, consistent with VBDS (arrow). VBDS: vanishing bile duct syndrome. |
Treatment
The treatment of VBDS is anchored in the management of the causative process. In the case of our patient, the suspected etiology was recurrence of classical HL. Nonetheless, the treatment proved difficult as many of the active therapies are contraindicated in acute hepatic failure, as their metabolic byproducts are hepatically metabolized. A decision was made to start the patient on ursodeoxycholic acid and cholestyramine. As a result of the therapy a minimal improvement in his laboratory values was observed. Subsequently, a pulse of high-dose steroids, dexamethasone 40 mg daily was initiated as means of controlling lymphoma progression and allowing for hepatic recovery. The patient was planned to receive a slow taper over several weeks to allow for hepatic recovery.
Follow-up and outcomes
The patient remained hospitalized for a period but wanted to seek a second opinion. He was discharged in stable clinical conditions and continued his care at a different academic institution.
| Discussion | ▴Top |
The diagnosis of VBDS in HL patients is challenging, as it is an extremely rare condition. Liver biopsy is required to make the diagnosis. Further, the initial presentation of the patient may be nonspecific without overt signs of recurrent lymphoma on physical exam or imaging. Review of reported cases of VBDS in the context of HL demonstrates that the most common presenting signs/symptoms (in order from most common to least common) are jaundice, vague epigastric/abdominal pain, diarrhea/loose stools, weight loss, low-grade/intermittent fevers, pruritus, and less commonly, nausea/vomiting and dark/tea-colored urine. HL defining B-symptoms at or preceding presentation is exceptionally rare and has been reported in a single case study [13]. In several studies, there was no evidence of HL at initial presentation on both exam and imaging studies, and the cause of VBDS was unknown until repeat imaging or physical exam revealed enlarged lymph nodes that were biopsied and ultimately revealed a diagnosis of de novo or recurrent HL [18-22]. In one case, the patient was diagnosed with HL after biopsy of lymph nodes that were discovered during screening imaging studies for liver transplantation [20, 22].
VBDS is a diagnosis of exclusion and requires that the clinicians rule out other causes of cholestasis, such as alcoholic or viral cirrhotic liver disease, gall stone disease (e.g., choledocholithiasis/ascending cholangitis), cancers of the biliary tree, and autoimmune diseases (e.g., PBC/PSC), among many others. Additionally, a liver biopsy must be performed with findings that meet strict criteria for the diagnosis of VBDS; mainly, ductopenia defined as a loss of interlobular bile ducts in more than 50% of portal tracts, with at least 11 portal tracts required for analysis (though 20 or more is deemed ideal) [1, 2, 23, 24]. These diagnostic criteria are meant to help distinguish VBDS from IC, which has also been reported as a consequence of HL. IC pathologic findings include cholestasis with inflammatory infiltrates and no significant ductopenia [25]. The relationship between VBDS and IC is unclear though it has been theorized that IC may either be a precursor to VBDS or that IC and VBDS are at opposite ends of a continuum of the same disease. A relationship between the two entities is supported by case reports of patients with HL and VBDS, where multiple liver biopsies have been taken throughout the disease course. Hallen et al reported the case of a patient that was ultimately diagnosed with VBDS from HL [19]. Initial liver biopsy showed “acute cholestasis and secondary toxic effects” but no ductopenia. A second liver biopsy 3 months later showed acute cholestasis and centrilobular necrosis, but again no loss of bile ducts. Finally, a third biopsy a short time later revealed severe ductopenia, and a diagnosis of VBDS was finally made. Similar findings in other serial biopsy studies have been observed with IC preceding the development of full-fledged VBDS [18].
The cause of ductopenia in patients with VBDS in the context of HL is still unknown. Previously, duct destruction and cholestasis were attributed to direct invasion of the hepatic parenchyma and bile ducts by lymphoma cells, but pathologic examination of specimens from these patients has failed to reveal any evidence of lymphoma infiltration. At present, the leading mechanistic theory is that HL cells secrete a cytokine that is either directly cytotoxic to bile duct epithelium or initiates immune system activation causing autoimmune destruction of bile duct epithelium. There are data suggesting that biliary epithelial cells express major histocompatibility complex (MHC) and intercellular adhesion molecule 1 (ICAM-1) in response to cytokines in liver allografts undergoing rejection, making them increasingly prone to immune-mediated destruction [26]. An autoimmune mechanism is further supported by the fact that biopsies from patients with VBDS have often shown immune infiltrates [1, 13, 22, 27, 28]. Similar cytokine-mediated processes have been suggested as the cause for cholestasis in other cancers, such as Stauffer syndrome with renal cell carcinoma that has been associated with increased interleukin (IL)-6 levels [25, 29].
There are limited data to explain why only a small subset of patients with HL develop VBDS. It has been proposed that this predisposition may be secondary to a host of genetic factors. Bakhit et al published a case report of a patient with HL and VBDS in whom genetic sequencing (via next-generation whole exome sequencing) was performed to look for molecular defects in genes related to bile acid transport or synthesis and identified a defect in the MST1 gene locus, a locus that is associated with both PSC and inflammatory bowel disease (IBD) [22]. This is even more interesting considering that several patients with VBDS and HL have been co-diagnosed with IBD/colitis [19-22]. This suggests that both VBDS and IBD/colitis in some HL patients may be due to a common paraneoplastic mechanism that is influenced by specific host genetic factors. Moreno et al studied 24 otherwise healthy adults with elevated GGT of unknown cause with liver biopsy [27]. They found that only 62% of their portal tracts had bile ducts. Repeat biopsy on three of these patients years later showed no change, including no worsening. Thus, some authors have theorized that patients that develop VBDS from HL may have baseline cholestasis and are thus more prone to VBDS than other patients with HL with normal bile duct structure/function at baseline [30].
The management of patients with VBDS secondary to HL is extremely challenging as the cholestasis and liver function abnormalities preclude full-dose first-line chemotherapeutic treatment with ABVD, Stanford V or BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone). Further, liver transplantation is not possible as active Hodgkin disease disqualifies patients for transplant eligibility. Nonetheless, treatment of the underlying HL is the only path forward in the management of patients with VBDS from HL, as left untreated VBDS progresses to biliary cirrhosis, liver failure, and subsequent death. Unfortunately, due to the rarity of the disease, the ideal strategy for treatment of patients with VBDS and HL is not known. As such, clinicians have employed a variety of strategies for treatment of HL in patients with VBDS including definite radiation therapy alone without chemotherapy, single agent chemotherapy with non-hepatically cleared agents (e.g., single agent mustard therapy with mechlorethamine), or modified/alternate regimens where hepatically cleared agents are dose-reduced or omitted (e.g., mechlorethamine, vincristine, procarbazine, and prednisone (MOPP)) [13, 18, 20-22, 28, 30, 31]. In some cases, these therapies have been used initially to try to reduce HL disease burden and alleviate VBDS/cholestasis so that standard treatment regimens (e.g., ABVD) can then be used upon resolution of hyperbilirubinemia [13, 17].
Despite prior beliefs that development of VBDS in patients with HL was irreversible, resolution of hepatic dysfunction and cholestasis has been seen with treatment and remission of the underlying HL [13, 17-19, 28]. Ballonoff et al reviewed all published cases of VBDS and IC and evaluated outcomes of these patients [31]. They found that 1-year overall survival and liver failure-free survival were 43% and 41%, respectively. Further, in the 20 patients with no evidence of residual HL after treatment, 60% achieved liver failure-free survival. While promising, this result reveals that even with treatment and remission of HL, liver failure and death may be unavoidable in a significant portion of patients. On univariate analysis, stage I/II HL, with complete response of HL to treatment, and radiation therapy were associated with statistically significant improved liver failure-free survival. However, it should be noted that this analysis grouped both patients with IC and VBDS together for outcomes. This was justified by the fact that pathologic diagnosis (IC vs. VBDS) was not associated with different outcome on univariate analysis (hazard ratio (HR) 0.81, 95% confidence interval (CI): 0.35 - 1.83); however, most IC cases (12 of 18) were pulled from before 1993 (before VBDS was a known entity), and thus many of these cases of IC may have been VBDS. Therefore, outcomes of patients with VBDS and HL may indeed be even worse than reported.
The treatment of our patient was additionally complicated by the fact that he had new onset and severe renal impairment in addition to marked direct hyperbilirubinemia and hepatic dysfunction. Because of this, treatment with small molecule chemotherapeutics, even modified regimens with reduced dose, was felt unsafe. The mechanism of our patient’s renal dysfunction is not known, but several other case reports of HL and VBDS have reported new onset renal dysfunction with reports of acute kidney injury (AKI) of unclear etiology and nephrotic syndrome [13, 22, 25]. Such cases need to be evaluated and treated in academic centers which have the experience and capability to manage such complex patients.
Conclusions
VBDS is a rare complication of HL that portends a poor prognosis. Without treatment, patients progress to biliary cirrhosis, liver failure, and death. As such, VBDS should remain on the differential diagnosis for patients with HL and unexplained jaundice. The underlying mechanism of VBDS in patients with HL remains unknown but is likely a paraneoplastic phenomenon related to cytokine release from lymphoma cells. Exclusion of other cholestatic processes and liver biopsy are required for the diagnosis of VBDS. Treatment of patients with VBDS in the context of HL should focus on treating the underlying HL to prevent worsening liver damage and hepatic failure. While some patients experience complete recovery of liver function with treatment, a significant proportion who receive treatment and achieve HL remission have persistent liver abnormality and may still progress to liver failure and death. Early diagnosis and treatment of underlying HL are likely crucial to improving outcomes in these patients.
Learning points
VBDS is a known complication of HL.
Recognition of VBDS requires a high index of clinical suspicion.
Diagnosis of VBDS is based on clinical presentation, as well as radiologic and pathologic evidence of this phenomenon.
Treatment includes management of the underlying HL which can be complicated by concurrent presence of hepatic failure. Supportive therapy including use of high-dose steroids and ursodeoxycholic acid are appropriate lines of therapy.
Acknowledgments
None to declare.
Financial Disclosure
The authors of this publication do not have any relevant financial disclosure.
Conflict of Interest
None to declare.
Informed Consent
Data used in this clinical case report were deidentified. Permission was granted to use for the data in educational publications.
Author Contributions
Olger Nano MD, FACP, as primary author of publication, assembled relevant clinical data as well as literature review, provided interpretation of clinical data and summary of relevant information in the literature. Stanislav Ivanov, MD, MPH, as editor, provided editing and formatting of publication. Taroon Kapoor, as editor, provided editing and formatting of publication.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
| References | ▴Top |
- Nakanuma Y, Tsuneyama K, Harada K. Pathology and pathogenesis of intrahepatic bile duct loss. J Hepatobiliary Pancreat Surg. 2001;8(4):303-315.
doi pubmed - Reau NS, Jensen DM. Vanishing bile duct syndrome. Clin Liver Dis. 2008;12(1):203-217.
doi pubmed - Kaseb H, Babiker HM. Cancer Lymphoma. Hodgkin, StatPearls. Treasure Island (FL). 2018
- Ansell SM. Hodgkin lymphoma: 2018 update on diagnosis, risk-stratification, and management. Am J Hematol. 2018;93(5):704-715.
doi pubmed - Roth A, Kolaric K, Dominis M. Histologic and cytologic liver changes in 120 patients with malignant lymphomas. Tumori. 1978;64(1):45-53.
doi pubmed - Jaffe ES. Malignant lymphomas: pathology of hepatic involvement. Semin Liver Dis. 1987;7(3):257-268.
doi pubmed - Colby TV, Hoppe RT, Warnke RA. Hodgkin's disease: a clinicopathologic study of 659 cases. Cancer. 1982;49(9):1848-1858.
doi pubmed - Levitan R, Diamond HD, Craver LF. Jaundice in Hodgkin's disease. Am J Med. 1961;30:99-111.
doi pubmed - Givler RL, Brunk SF, Hass CA, Gulesserian HP. Problems of interpretation of liver biopsy in Hodgkin's disease. Cancer. 1971;28(5):1335-1342.
doi pubmed - Kim H, Dorfman RF, Rosenberg SA. Pathology of malignant lymphomas in the liver: application in staging. Prog Liver Dis. 1976;5:683-698.
pubmed - Hubscher SG, Lumley MA, Elias E. Vanishing bile duct syndrome: a possible mechanism for intrahepatic cholestasis in Hodgkin's lymphoma. Hepatology. 1993;17(1):70-77.
pubmed - Ghobrial IM, Wolf RC, Pereira DL, Fonseca R, White WL, Colgan JP, Habermann TM, et al. Therapeutic options in patients with lymphoma and severe liver dysfunction. Mayo Clin Proc. 2004;79(2):169-175.
doi pubmed - Sathyanarayanan V, Foo WC, Fanale M, Westin J. Deeper insights into vanishing bile duct syndrome in lymphoma: a perplexing entity. Clin Lymphoma Myeloma Leuk. 2016;16(5):e65-70.
doi pubmed - Lindor KD, Dickson ER, Baldus WP, Jorgensen RA, Ludwig J, Murtaugh PA, Harrison JM, et al. Ursodeoxycholic acid in the treatment of primary biliary cirrhosis. Gastroenterology. 1994;106(5):1284-1290.
doi pubmed - Poupon RE, Poupon R, Balkau B. Ursodiol for the long-term treatment of primary biliary cirrhosis. The UDCA-PBC Study Group. N Engl J Med. 1994;330(19):1342-1347.
doi pubmed - Kondrackiene J, Beuers U, Kupcinskas L. Efficacy and safety of ursodeoxycholic acid versus cholestyramine in intrahepatic cholestasis of pregnancy. Gastroenterology. 2005;129(3):894-901.
doi pubmed - Rota Scalabrini D, Caravelli D, Carnevale Schianca F, D'Ambrosio L, Tolomeo F, Boccone P, Manca A, et al. Complete remission of paraneoplastic vanishing bile duct syndrome after the successful treatment of Hodgkin's lymphoma: a case report and review of the literature. BMC Res Notes. 2014;7:529.
doi pubmed pmc - Crosbie OM, Crown JP, Nolan NP, Murray R, Hegarty JE. Resolution of paraneoplastic bile duct paucity following successful treatment of Hodgkin's disease. Hepatology. 1997;26(1):5-8.
doi pubmed - Hallen K, Sangfelt P, Nilsson T, Nordgren H, Wanders A, Molin D. Vanishing bile duct-like syndrome in a patient with Hodgkin lymphoma - pathological development and restitution. Acta Oncol. 2014;53(9):1271-1275.
doi pubmed - Bakhit M, McCarty TR, Park S, Njei B, Cho M, Karagozian R, Liapakis A. Vanishing bile duct syndrome in Hodgkin's lymphoma: a single center experience and clinical pearls. J Clin Gastroenterol. 2016;50(8):688.
doi pubmed pmc - DeBenedet AT, Berg CL, Enfield KB, Woodford RL, Bennett AK, Northup PG. A case of vanishing bile duct syndrome and IBD secondary to Hodgkin's lymphoma. Nat Clin Pract Gastroenterol Hepatol. 2008;5(1):49-53.
doi pubmed - Bakhit M, McCarty TR, Park S, Njei B, Cho M, Karagozian R, Liapakis A. Vanishing bile duct syndrome in Hodgkin's lymphoma: A case report and literature review. World J Gastroenterol. 2017;23(2):366-372.
doi pubmed pmc - Ludwig J, Wiesner RH, LaRusso NF. Idiopathic adulthood ductopenia. A cause of chronic cholestatic liver disease and biliary cirrhosis. J Hepatol. 1988;7(2):193-199.
doi pubmed - Ludwig J, Wiesner RH, Batts KP, Perkins JD, Krom RA. The acute vanishing bile duct syndrome (acute irreversible rejection) after orthotopic liver transplantation. Hepatology. 1987;7(3):476-483.
doi pubmed - Nader K, Mok S, Kalra A, Harb A, Schwarting R, Ferber A. Vanishing bile duct syndrome as a manifestation of Hodgkin's lymphoma: a case report and review of the literature. Tumori. 2013;99(4):e164-168.
doi pubmed - Adams DH, Hubscher SG, Shaw J, Rothlein R, Neuberger JM. Intercellular adhesion molecule 1 on liver allografts during rejection. Lancet. 1989;2(8672):1122-1125.
doi pubmed - Moreno A, Carreno V, Cano A, Gonzalez C. Idiopathic biliary ductopenia in adults without symptoms of liver disease. N Engl J Med. 1997;336(12):835-838.
doi pubmed - Leeuwenburgh I, Lugtenburg EP, van Buuren HR, Zondervan PE, de Man RA. Severe jaundice, due to vanishing bile duct syndrome, as presenting symptom of Hodgkin's lymphoma, fully reversible after chemotherapy. Eur J Gastroenterol Hepatol. 2008;20(2):145-147.
doi pubmed - Blay JY, Rossi JF, Wijdenes J, Menetrier-Caux C, Schemann S, Negrier S, Philip T, et al. Role of interleukin-6 in the paraneoplastic inflammatory syndrome associated with renal-cell carcinoma. Int J Cancer. 1997;72(3):424-430.
doi pubmed - de Medeiros BC, Lacerda MA, Telles JE, da Silva JA, de Medeiros CR. Cholestasis secondary to Hodgkin's disease: report of 2 cases of vanishing bile duct syndrome. Haematologica. 1998;83(11):1038-1040.
pubmed - Ballonoff A, Kavanagh B, Nash R, Drabkin H, Trotter J, Costa L, Rabinovitch R. Hodgkin lymphoma-related vanishing bile duct syndrome and idiopathic cholestasis: statistical analysis of all published cases and literature review. Acta Oncol. 2008;47(5):962-970.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.