

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 14, Number 9-10, October 2023, pages 327-331

A Double Hit to Ubiquitination Leading to a New Diagnosis of VEXAS Syndrome
Helen Pozdniakovaa, c ![]() , Apurva Vedirea, Anand Kadakiaa, Steven Imburgioa, Ravneet Bajwab, Varsha Guptab, Ruchi Bhattb, Mohammad A. Hossaina
, Apurva Vedirea, Anand Kadakiaa, Steven Imburgioa, Ravneet Bajwab, Varsha Guptab, Ruchi Bhattb, Mohammad A. Hossaina
aDepartment of Internal Medicine, Jersey Shore University Medical Center, Neptune, NJ, USA
bDepartment of Hematology Oncology, Jersey Shore University Medical Center, Neptune, NJ, USA
cCorresponding Author: Helen Pozdniakova, Department of Internal Medicine, Jersey Shore University Medical Center, Neptune, NJ 07753, USA
Manuscript submitted June 9, 2023, accepted July 8, 2023, published online October 13, 2023
Short title: A Doubt Hit to Ubiquitination: VEXAS Syndrome
doi: https://doi.org/10.14740/jmc4127
| Abstract | ▴Top |
VEXAS (vacuoles, E1 enzyme, X-linked, auto-inflammatory, somatic) syndrome is a newly defined illness that bridges hematology, oncology, and rheumatology. Its pathophysiology originates in a mutation in the UBA1 gene that leads to a defect in ubiquitination resulting in a severe systemic inflammatory syndrome. It is associated with significant morbidity and mortality; however, data are scarce due to limited cases described in the literature. Here we describe a case of a male in his 60s who was referred to hematology-oncology due to progressive dyspnea, poor oral intake, and weight loss. He was diagnosed with relapsing polychondritis 2 years prior; however, his symptoms did not improve despite treatment. He was ultimately diagnosed with VEXAS syndrome with a mutation in UBA1 (ubiquitin-like modifier activating enzyme 1) and a concurrent SQSTM1 mutation. In addition, the coexistence of two mutations in the ubiquitination pathway in the same patient has not been reported to date. This patient and the treatment course were compared to pre-existing literature to increase awareness and improve the medical management of VEXAS syndrome.
Keywords: VEXAS syndrome; Relapsing polychondritis; Myelodysplastic syndrome; X-linked; SQSTM1; UBA1; Anemia; Macrocytosis
| Introduction | ▴Top |
VEXAS (vacuoles, E1 enzyme, X-linked, auto-inflammatory, somatic) syndrome is a somatic mutation in the UBA1 (ubiquitin-like modifier activating enzyme 1) gene primarily affecting hematopoietic progenitor cells that often manifests in adulthood [1]. The main clinical features of VEXAS syndrome include a late onset treatment refractory inflammatory syndrome, often relapsing polychondritis (RP) with associated hematological abnormalities, most often occurring in men. UBA1 is an X-linked gene encoding the ubiquitin-activating enzyme 1, which is necessary for activating ubiquitylation of proteins involved in the autophagy-lysosome system [1, 2]. This syndrome is often severe and progressive and bridges both rheumatology and hematology due to its systemic inflammatory involvement. Early identification can lead to enhanced quality of life for our patients [1].
Here we investigate a unique case of a patient with concurrent UBA1 and SQSTM1 mutations in a patient with a severe and protracted illness course. This report discusses the pathophysiology of these mutations and the clinical implications in patients with VEXAS syndrome.
| Case Report | ▴Top |
Investigations
A male in his late 60s with a past medical history significant for rheumatoid arthritis presented with a long history of diffuse arthritic pain, shortness of breath, chronic ageusia, weight loss, and chronic transfusion-dependent anemia. His shortness of breath was so severe that he was unable to complete his activities of daily living. He was diagnosed with RP 2 years prior, however, despite treatment his symptoms progressed. Prior to admission, he was prescribed prednisone 20 mg daily, methotrexate 20 mg weekly, and periodic packed red blood cell transfusions for symptomatic anemia. The vitals of the presentation were unremarkable. Physical exam findings were significant for a frail-appearing male with right-sided conjunctivitis, inspiratory wheezing, and bilateral arm hyperpigmentation with skin atrophy.
Diagnosis
Lab values are presented in Table 1. At the time of presentation, his complete blood count was significant for leukocytosis with lymphopenia, elevated metamyelocytes, myelocytes, and promyelocytes. In addition, he had macrocytic anemia with red blood cell morphology that showed slight micro- and macrocytosis, schistocytes, and polychromasia. The workup of other etiologies (Table 2) was unremarkable including leukemia flow cytometry, glucose-6-phosphate dehydrogenase (G6PD), and paroxysmal nocturnal hemoglobinuria. Autoimmune workup was significant for a negative antinuclear antibody but elevated thyroid peroxidase and ribonucleoprotein antibodies. Of note, perinuclear anti-neutrophil cytoplasmic antibodies (P-ANCA) titer was 1:160. Due to his myelocyte predominant differential, he underwent a bone marrow biopsy which showed a hypercellular bone marrow with vacuoles, 5% blasts, and no evidence of dyspoiesis (Fig. 1a, b). The next generation sequencing and fluorescence in situ hybridization for multiple genetic abnormalities in the myelodysplastic syndrome (MDS) panel was negative. Some of the key mutations tested were genes such as FLT3, IDH1, IDH2, and chromosomal abnormalities such as del-5q, del-7q, monosomy 5, monosomy 7, trisomy 8, etc. Flow cytometry showed left shifted myeloid maturation pattern with 4.4% myeloblasts. Ultimately, he tested positive for a heterozygous UBA1 mutation at p.Met41Thr, which was consistent with the diagnosis of VEXAS syndrome. In addition, the patient was found to have a concurrent germline mutation in SQSTM1 at p.Pro392Leu.
 Click to view | Table 1. Initial Laboratory Values |
Click to view | Table 2. Autoimmune Workup |
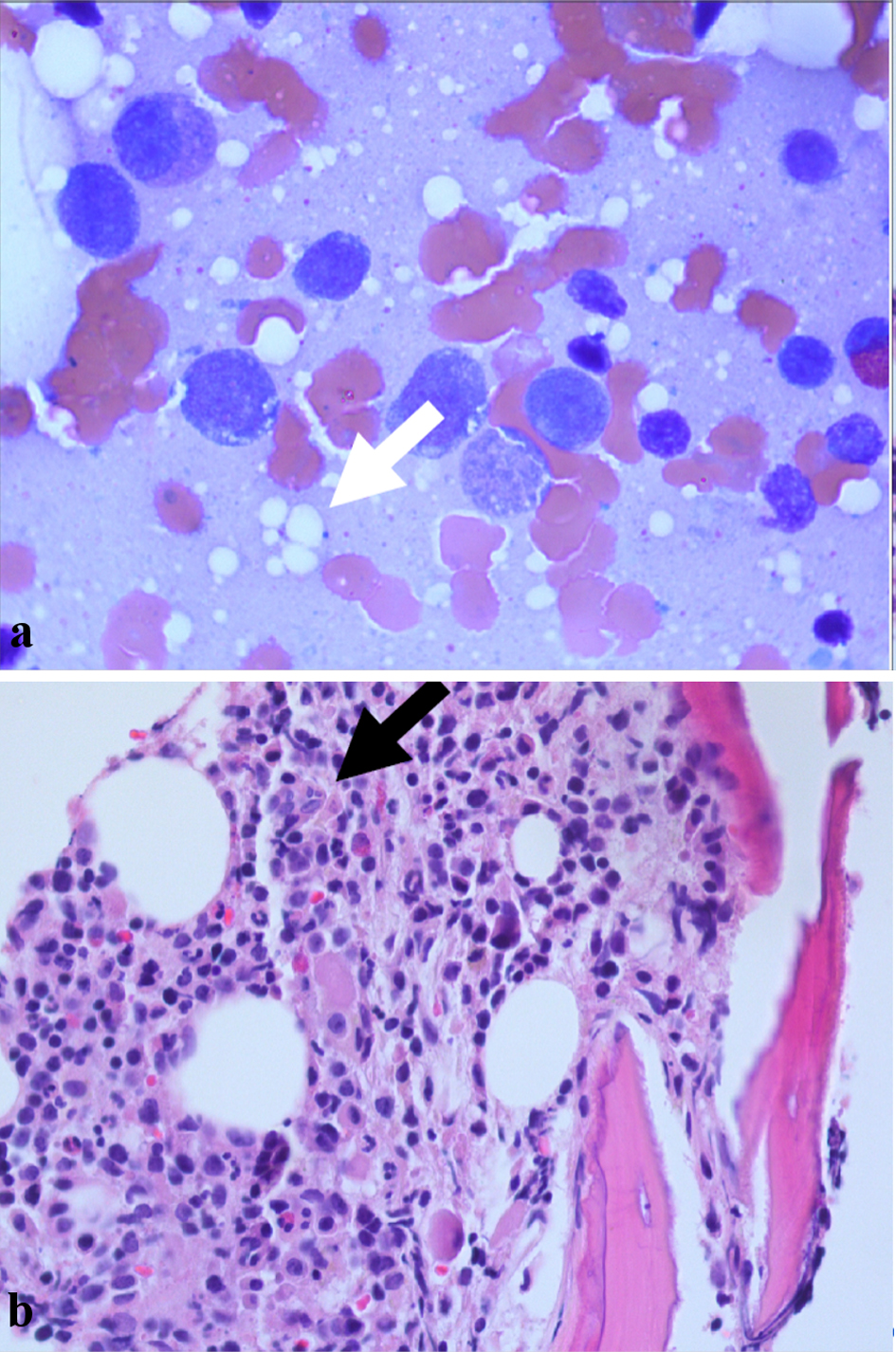 Click for large image | Figure 1. (a) Bone marrow biopsy showing numerous precursor cells including metamyelocytes and promyelocytes with intracellular vacuoles (white arrow). (b) Bone marrow biopsy showing hypercellular bone marrow (black arrow). |
Treatment
He was treated with prednisone 20 mg daily. Given the prolonged duration of steroid treatment, he required prophylaxis with fluconazole, trimethoprim-sulfamethoxazole, and acyclovir. For his chronic pain secondary to his multifactorial arthritis, he required numerous modalities of pain control including opioids. For the anemia, he received epoetin alfa injections every 2 weeks and red blood cell transfusions as needed.
Follow-up and outcomes
He was referred for an alloimmune bone marrow transplant and is awaiting a donor at this time.
| Discussion | ▴Top |
In this case, we described our patient who had a severe and protracted illness course that ultimately led to the diagnosis of VEXAS syndrome by genetic analysis. In addition, he was found to have a concurrent mutation in the ubiquitination pathway in the SQSTM1 gene which we hypothesize contributed to his severe presentation. Given the benefit early diagnosis can have on treatment options and quality of life, we hope to shed more light on this rare disease to improve awareness and diagnosis for patients such as ours.
Initial identification of VEXAS syndrome was initially published in a New England Journal of Medicine article in 2020 by Beck et al [3]. This study included 25 patients, 100% of whom were male with a median age of 64 and all were identified to have a somatic UBA1 variant. Key clinical features on presentation included fever in 92%, skin involvement in 88%, and pulmonary infiltrates in 72% [3]. The patient in our case was male (consistent with the X-linked recessive pattern) and initially presented with evidence of skin involvement and chronic shortness of breath [2]. Hematologically, the most common presentation was macrocytic anemia in 96%, and all patients were found to have bone marrow vacuoles which were present in our case. Analysis of isolates from the bone marrow showed abundant mutant granulocytes, megakaryocytes, and lymphoid progenitors but very few mutated mature lymphocytes [3]. In addition, patients in this study had decreased peripheral lymphocyte counts which suggested that lymphocytes were likely not able to proliferate if they had this mutation or were eliminated. Identification of the UBA1 mutation was a critical finding and it is thought to be the primary driver of the symptoms of VEXAS syndrome [3]. Specifically, the p.Met41 mutation was identified as a somatic mutation and was distributed as a mosaic pattern affecting different cell types suggesting that this mutation is likely lethal as germline and only compatible with life when manifesting in specific cell types [3]. The SQSTM1 gene encodes the scaffolding protein p62, which is responsible for nuclear factor-kappa B (NF-kappa B) signaling [4, 5]. To our knowledge, the coexistence of the UBA1 and SQSTM1 mutations in the same patient has not been reported to date.
Previous studies have identified that approximately 60% of patients with VEXAS syndrome meet the criteria for RP which is diagnosed either by McAdam’s or Damiani’s diagnostic criteria [6, 7], which was the initial presentation of the patient in our case report. Among patients with RP, male sex, and a mean corpuscular volume > 100 fL or platelet count < 200 × 109/L predicted VEXAS syndrome with near-perfect accuracy [2]. It is important to identify patients with VEXAS that have RP because it may be a marker of a more severe disease [7]. A National Institutes of Health study published in the Academy of Rheumatology looked at 98 patients and made some key observations. Compared to patients diagnosed with pure RP, VEXAS was often refractory to medications other than glucocorticoids and received on average a higher amount of glucocorticoid use [8]. In addition, VEXAS patients had higher maximum erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) levels, greater prevalence of detectable lupus anticoagulant, rheumatoid factor, thrombocytopenia, anemia, and macrocytosis [8]. In the study by Beck et al [3], of the 25 patients analyzed, 100% had current or past treatment with glucocorticoids and presented with a mean CRP of 73 and ESR of 97.
Importantly VEXAS syndrome has been found to have a high frequency of secondary MDS diagnosed anywhere in 25-55% of patients [2]. It is unclear if the UBA1 mutation is a driver mutation or if chronic inflammation it induces allows for the development of MDS [2]. Although our case had evidence of mildly increased blasts in both the bone marrow and peripheral blood, his bone marrow biopsy was ultimately negative by flow cytometry for any pathologic mutations. It is clear that early identification of this syndrome and bone marrow biopsy is an important part of management [2].
Clear guidelines for the treatment of VEXAS syndrome have not yet been defined. Most patients are treated with high-dose glucocorticoids, with additional steroid-sparing agents as well [2]. Glucocorticoids generally exert their anti-inflammatory effects by direct binding of the glucocorticoid or glucocorticoid receptor complex to areas in the promoter region of genes that are responsive to them. Alternatively, this complex can interact with other transcription factors such as activating protein-1 or NF-kappa B [9, 10] The mechanism by which glucocorticoids seem to have such a strong beneficial effect in VEXAS syndrome is likely through suppression of the NF-kappa B activation. Through the induction of I kappa B alpha inhibitory protein, the activated NF-kappa B can be confined inside inactive cytoplasmic complexes [10]. The exact mechanism through which UBA1 mutation causes the disease is still a matter of discussion. One suggested pathway is due to the activation of the unfolded protein response and of multiple inflammatory pathways including interferon-a and interferon-b pathways [11]. This is the reason that Janus Kinase inhibitors are gaining popularity as a treatment option, as they inhibit Janus kinases which are downstream effectors in interferon signaling [12]. NF-kappa B is a transcription factor that regulates immune responses and also plays a role in inflammatory pathways. Its activity is regulated by phosphorylation and ubiquitylation. As discussed above, VEXAS syndrome is due to pathogenic variants in UBA1, which is the enzyme involved in cellular ubiquitylation [12]. So perhaps the resulting defects in ubiquitylation could affect NF-kappa B pathways, which are remedied by glucocorticoid activity. Hypomethylating agents like azacytidine have also been used as treatment options [7]. In patients refractory to multiple therapies, allogeneic hematopoietic stem cell transplantation (ASCT) is certainly a therapeutic option. Although clinical trials have yet to be performed, successful case reports of patients treated with ASCT have been recently published [13].
In conclusion, VEXAS syndrome is a newly discovered entity that when diagnosed early, can have a large impact on a patient’s quality of life and treatment options. Further investigation needs to be done regarding the significance of a concurrent SQSTM1 mutation that affects the same pathway and may be linked to more severe symptoms. Given VEXAS’s association with MDS, early identification can facilitate earlier testing and possibly improved outcomes. Lastly, treatment options are limited and require further investigation, but stem cell transplant has shown to be promising.
Learning points
VEXAS syndrome is a newly identified syndrome that bridges rheumatology and hematology and oncology, resulting in systemic inflammation, various blood abnormalities, and often RP.
Multiple mutations in the ubiquitination pathway may contribute to a more severe disease.
Early identification of this syndrome can lead to improved quality of life and open the door for new treatment modalities for our patients.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
The authors declare that there is no conflict of interests regarding the publication of this paper.
Informed Consent
The patient described in the case reports had given informed consent for the case report to be published.
Author Contributions
Each author was individually involved in and made substantial contributions to conceptions and designs, acquisition of data, analysis, interpretation of data, drafting, and editing the manuscript. HP and AV contributed to the designs, acquisition of data, analysis, interpretation of data, drafting, and editing of the manuscript. AK and SI contributed to the drafting and editing of the manuscript. R. Bajwa and VG contributed to the designs and analysis of the manuscript. R. Bhatt and MH contributed to the designs, analysis, interpretation of data, and editing of the manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
VEXAS: vacuoles, E1 enzyme, X-linked, auto-inflammatory, somatic; UBA1: ubiquitin-like modifier activating enzyme 1; MDS: myelodysplastic syndrome; NF-kappa B: nuclear factor-kappa B activation
| References | ▴Top |
- Marcela Ferrada. VEXAS syndrome. National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIAMS. 2022. https://www.niams.nih.gov/labs/grayson-lab/vexas.
- Grayson PC, Patel BA, Young NS. VEXAS syndrome. Blood. 2021;137(26):3591-3594.
doi pubmed pmc - Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, Balanda N, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med. 2020;383(27):2628-2638.
doi pubmed pmc - Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171(4):603-614.
doi pubmed pmc - Daroszewska A, van 't Hof RJ, Rojas JA, Layfield R, Landao-Basonga E, Rose L, Rose K, et al. A point mutation in the ubiquitin-associated domain of SQSMT1 is sufficient to cause a Paget's disease-like disorder in mice. Hum Mol Genet. 2011;20(14):2734-2744.
doi pubmed - Sosada B, Loza K, Bialo-Wojcicka E. Relapsing polychondritis. Case Rep Dermatol Med. 2014;2014:791951.
doi pubmed pmc - Ferrada MA, Sikora KA, Luo Y, Wells KV, Patel B, Groarke EM, Ospina Cardona D, et al. Somatic mutations in UBA1 define a distinct subset of relapsing polychondritis patients with VEXAS. Arthritis Rheumatol. 2021;73(10):1886-1895.
doi pubmed - Obiorah IE, Patel BA, Groarke EM, Wang W, Trick M, Ombrello AK, Ferrada MA, et al. Benign and malignant hematologic manifestations in patients with VEXAS syndrome due to somatic mutations in UBA1. Blood Adv. 2021;5(16):3203-3215.
doi pubmed pmc - van der Velden VH. Glucocorticoids: mechanisms of action and anti-inflammatory potential in asthma. Mediators Inflamm. 1998;7(4):229-237.
doi pubmed pmc - Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science. 1995;270(5234):286-290.
doi pubmed - Beck DB, Werner A, Kastner DL, Aksentijevich I. Disorders of ubiquitylation: unchained inflammation. Nat Rev Rheumatol. 2022;18(8):435-447.
doi pubmed pmc - Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7(8):758-765.
doi pubmed pmc - Diarra A, Duployez N, Fournier E, Preudhomme C, Coiteux V, Magro L, Quesnel B, et al. Successful allogeneic hematopoietic stem cell transplantation in patients with VEXAS syndrome: a 2-center experience. Blood Adv. 2022;6(3):998-1003.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.