

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 14, Number 9-10, October 2023, pages 339-343

A Case Report of a Challenging Disease: Immunoglobulin G4-Related Disease With Acute Kideny Injury
Mostafa Mohraga, d ![]() , Mohammed Abdulrasakb, c, Mohammed Binsalmanb, c, Majid Darraja
, Mohammed Abdulrasakb, c, Mohammed Binsalmanb, c, Majid Darraja
aDepartment of Medicine, Faculty of Medicine, Jazan University, Jazan, Saudi Arabia
bDepartment of Clinical Sciences, Malmo, Lund University, Malmo, Sweden
cDepartment of Gastroenterology and Nutrition, Skane University Hospital, Malmo, Sweden
dCorresponding Author: Mostafa Mohrag, Department of Medicine, Faculty of Medicine, Jazan University, Jazan, Saudi Arabia
Manuscript submitted September 16, 2023, accepted October 9, 2023, published online October 13, 2023
Short title: IgG4-RD With Acute Kidney Injury
doi: https://doi.org/10.14740/jmc4159
| Abstract | ▴Top |
Immunoglobulin G4-related disease (IgG4-RD), which was initially identified as a type of autoimmune pancreatitis around the year 2000, is now widely acknowledged to be a systemic sickness. Based on both general and organ-specific criteria, alongside laboratory measurements of IgG4-subtype, the diagnosis is made. The diagnosis requires, however, a heightened index of suspicion, especially given the nonspecific clinical presentation. In addition to this, the symptoms may be “disseminated” in time and the multitude of organ-system involvement may seem initially unrelated. Furthermore, IgG4 levels may be falsely normal especially during the first presentation of IgG4-RD. We report a case of a 33-year-old male who was referred by his general practitioner (GP) to the fast access nephrology clinic due to elevated creatinine and fatigue, which was found after the patient had undergone some investigations at the GP office. He had history of atopic dermatitis and a prior admission for acute pancreatitis of unknown cause and recent bilateral anterior uveitis treated with steroid eyedrops. His urinalysis showed one to two granular casts per high-power field (HPF), and his creatinine was 262 µmol/L (previously normal). Three main differential diagnoses were considered given the patient’s history: sarcoidosis, tubulointerstitial nephritis with uveitis (TINU) and IgG4-related disorder. Investigations were undertaken in that regard showing elevated serum IgG4 levels (2.7 times upper-limit of normal). Renal biopsy demonstrated tubulointerstitial nephritis (TIN) with 30 IgG4-positive plasma cells per HPF. Given the patient’s presentation over time, a diagnosis of IgG4-TIN was considered. The patient was treated with high-dose steroids and has shown signs of improvement of both his renal and ocular problems. The uniqueness of the case is reflected through the fact that IgG4-renal disease is usually diagnosed in patients with an already established manifestation of another organ, whilst in our patient the renal involvement led to establishing IgG4-RD. It is also important to note that, in spite of initially negative serum IgG4 levels, the diagnosis still needs to be considered especially if multisystem involvement is present (as in this case).
Keywords: IgG4-related disease; IgG-TIN; Acute kidney injury
| Introduction | ▴Top |
Immunoglobulin G4-related disease (IgG4-RD) is a relatively recently described disorder that can affect multiple systems, either simultaneously or with a certain time gap in between the presentations. Clinically, the disease may have nonspecific symptoms, and delay to diagnosis is common, as it may affect any organ. It should, however, be suspected in patients with constitutional symptoms (e.g., weight loss, fevers, and tiredness) alongside multiple organ involvement which may be present on clinical exam, imaging or laboratory workup [1]. Such involvement includes (but is not limited to) organs such as kidneys, liver and biliary tract, pancreas and the retroperitoneum. The disease is more commonly present in patients with a prior history of atopic dermatitis or allergic rhinitis and asthma, suggesting potential autoimmune pathogenesis for this disorder. The common denominator for the organ involvement is the presence of elevated serum IgG4 levels, alongside the infiltration of that specific organ with IgG4-positive plasma cells [2].
Given the multitude of organs that IgG4-RD may involve, the diagnosis may be elusive. The commonest presentation of IgG4-RD is diagnosing it as part of investigation for unclear pancreatitis, with classic “sausage pancreas” finding on cross-sectional imaging alongside elevated serum IgG4 levels [2]. Other presentations of this rare disorder may include (but are not limited to) sclerosing cholangitis, ocular involvement or renal involvement, occurring simultaneously or dispersed over time in the same patient.
| Case Report | ▴Top |
Investigations
A 33-year-old man with a history of nephrolithiasis, atopic dermatitis since childhood treated with topical steroids, a bout of pancreatitis of unknown cause in December 2022, when total IgG and IgG4 levels were reported as normal, and bilateral anterior uveitis treated with steroid drops at the ophthalmology department in February 2023, presented to his general practitioner (GP) as part of a follow-up for his prior pancreatitis in early March 2023. Prior to the visit the patient had some laboratory tests performed, revealing an elevated serum creatinine (262 µmol/L), an elevated C-reactive protein (CRP; 59 mg/L) and erythrocyte sedimentation rate (ESR) (48 mm/h) and thrombocytosis (623 × 109). His previous creatinine had been normal. Therefore, he was referred to the fast-access renal clinic for assessment and further history.
The patient history was negative for the intake of nonsteroidal anti-inflammatory drugs (NSAIDs) or other potential nephrotoxic agents. The patient reported no known weight loss; however, he felt that his clothes have grown a bit looser. He also felt that his appetite had decreased slightly after the bout of pancreatitis. He reported unchanged thirst sensation and subjectively normal urine amount. He reported no discoloration or frothiness of the urine, no flank pain and no joint pain or swelling. His vision was getting better with the steroid drops. The patient had no family history of autoimmune disorders. The patient demonstrated a normal physical exam. Urine dipstick showed 1+ glucose, 1+ red blood cell (RBC), 2+ protein, 1+ white blood cell (WBC), negative for ketones and nitrites. Urinalysis showed one to two granular casts per high-power field (HPF), and one to two waxy cylinders per HPF. The patient’s weight was taken during the visit, and it was at 60 kg (previous weight 68 kg).
Diagnosis
Given the patient’s prior presentation with pancreatitis and being, at presentation, treated for bilateral anterior uveitis apart from newly-onset renal failure, three main differential diagnoses were entertained: multisystemic sarcoidosis, tubulointerstitial nephritis with uveitis (TINU) and IgG4-related syndrome. Repeat blood tests were taken with autoimmune screen (antinuclear antibody (ANA) and antineutrophil cytoplasmic antibody (ANCA)), complement C3 and C4 levels, serum angiotensin-converting enzyme (ACE) levels, and serum and urinary protein electrophoresis (SPEP and UPEP). The patient was admitted for renal biopsy, with the aforementioned differential diagnoses in mind. He underwent a non-enhanced computed tomography (CT) of chest and abdomen, revealing normal sized kidneys and sub-10 mm adenopathy in the paraaortic region and axillae (Fig. 1).
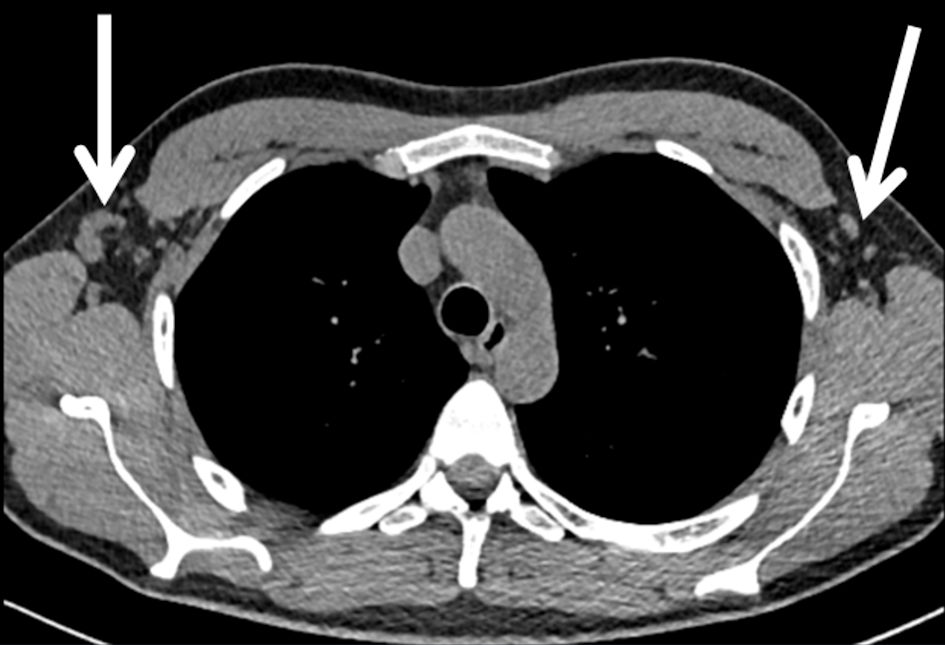 Click for large image | Figure 1. Subcentimeter adenopathy in bilateral axillae (arrows). |
While awaiting results of investigations, the patient underwent a renal biopsy. This was uncomplicated. The preliminary report showed typical findings of tubulointerstitial nephritis, with severe inflammation dominated by lymphocytes and some plasma cells with eosinophils. No granulomas were visualized in the tubulo-interstitium (Fig. 2).
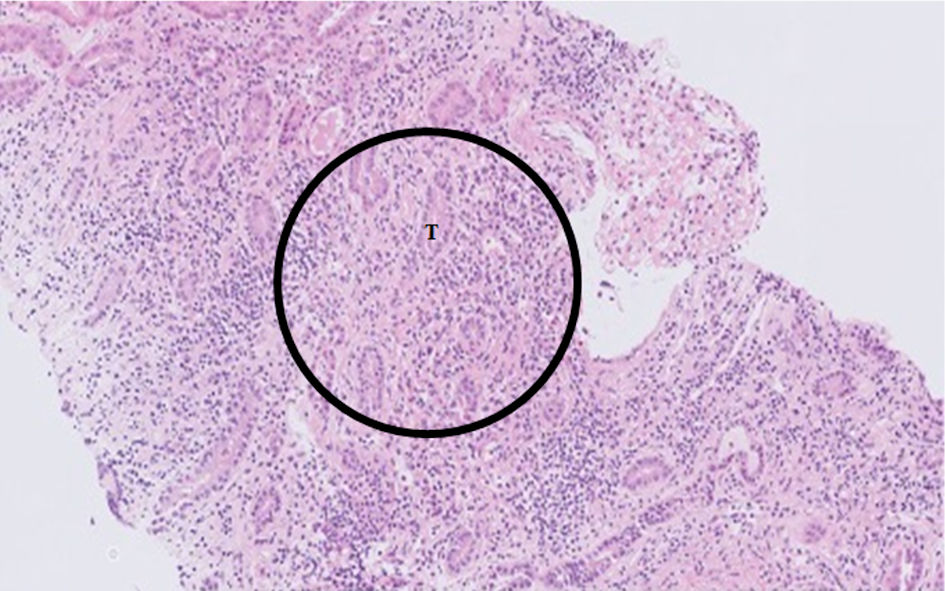 Click for large image | Figure 2. Hematoxylin and eosin (H&E) stain at × 10 magnification of renal biopsy specimen, showing diffuse lymphocytic infiltrate (circle) in the tubule-interstitium and surrounding tubules (T). |
While immunohistochemical analyses for CD3, CD20 and IgG4 markers, amongst others, were pending, the patient was promptly started on prednisolone 50 mg daily with planned taper of 2.5 mg weekly, with planned initial weekly follow-up of renal function tests.
The report of SPEP showed an elevated IgG level of 21.4 g/L, which was normal at the initial pancreatitis some months ago, and with UPEP showing tubular proteinuria with elevated protein HC/creatinine index (31 g/mol) with elevated urinary kappa/creatinine and lambda/creatinine ratio (20 g/mol and 8.8 g/mol) but without Bence-Jones proteinuria. Complement levels (C3 and C4 levels) were reported as normal, as well as serum ACE levels. Autoimmune screening was negative for ANA and ANCA antibodies.
IgG-subclasses were ordered, with IgG4 being elevated at 3.42 g/L (i.e., about 2.7 × upper limit of normal). The biopsy report showed up to 30 IgG4-positive plasma cells per 40 × magnified HPF, with CD3-positive infiltrates and some CD20-positive pockets, albeit lacking classic storiform fibrosis (Fig. 3).
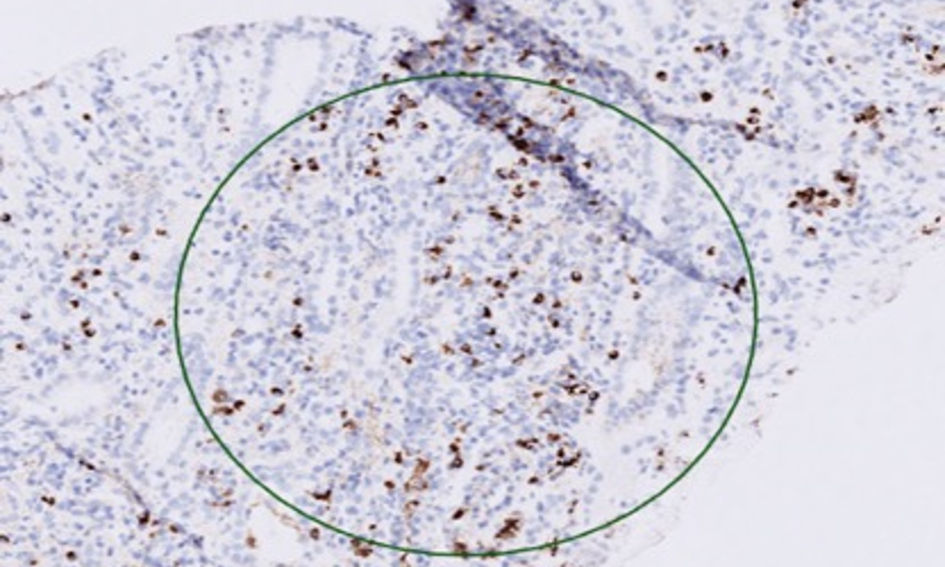 Click for large image | Figure 3. IgG4 immunohistochemical staining at × 10 magnification showing IgG4-positive lymphocytes in the tubulo-interstitium (circle). Ig: immunoglobulin. |
Hematopathologist input was sought given the absence of classic storiform fibrosis. The IgG4/IgG positive lymphocytes ratio was 50%, with up to 57 IgG4-positive cells/HPF (sized at 0.2 mm3). In spite of this, when applying the criteria as per the European League Against Rheumatism (EULAR) guidelines, there was a moderately dense lymphocytic infiltrate without fibrosis and obliterative thrombophlebitis, and thus there was uncertainty with regards to histopathological diagnosis of IgG4-RD. However, given the absence of an alternative diagnosis explaining all the patient’s findings, in addition to clearly elevated IgG4-levels, the diagnosis of IgG4-RD was established on a clinical basis.
Treatment
On the basis of the preliminary biopsy report, showing tubulointerstitial nephritis (TIN), the patient was started on prednisolone 50 mg daily with planned taper of 5 mg per week. When the diagnosis of IgG4-RD was established, the taper was slowed down to 2.5 mg weekly taper.
Follow-up and outcomes
Within a week of starting steroids, the patient’s creatinine had dropped to 133 µmol/L with successive normalization of inflammatory markers and IgG levels. The patient’s ocular symptoms also abated on starting systemic steroids.
| Discussion | ▴Top |
IgG4-RD is an immune-mediated fibroinflammatory disorder that often manifests as high blood IgG4 levels with certain histopathological features. It may affect several organs. IgG4-RD is a very novel phenomenon; hence we know very little about its etiology, prevalence, and epidemiology. Although it is believed that aberrant adaptive immune responses play a significant role in the development of IgG4-RD, the precise mechanisms are still not fully known. Notably, individuals with IgG4-RD frequently get allergic illnesses including atopic dermatitis [3, 4]. Most often affected areas include the lacrimal glands, salivary glands, orbital disease, retroperitoneum, lymph nodes, kidney, and lungs, while involvement of nearly all anatomical locations has been observed [5].
IgG4-RD is categorized as a fibroinflammatory illness, and both innate and adaptive immunological systems are involved in its pathogenesis. IgG4 antibodies are unique to humans and make up 1-4% of all IgG subtypes [6]. The precise etiology of IgG4-RD is still unknown. Certain susceptibility genes and environmental factors have been implicated [7].
Common pathological features are storiform fibrosis, obliterative phlebitis, mild to moderate eosinophilia, and thick lymphoplasmacytic infiltrates made up of IgG4+ plasma cells. A particularly distinctive finding is obliterative phlebitis, which is characterized by total or partial obliteration of venous veins and infiltration of the vessel wall or lumen with a dense lymphoplasmacytic infiltrate made up of lymphocytes and plasma cells [8, 9].
The most frequent form of IgG4-related kidney involvement is TIN. Investigations for suspected kidney masses, abnormal urine analysis, and/or renal failure may reveal the presence of IgG4-related TIN. The renal interstitium has lymphoplasmacytic infiltration, along with tubular atrophy and fibrosis, according to histological results. Low C3 and C4 levels are particularly common in patients with active illness in those with IgG4-related TIN. Since IgG4 only weakly binds to complement, it is unclear why hypocomplementemia develops in IgG4-TIN. Therefore, it is believed that IgG1 and IgG3 are responsible for hypocomplementemia. Enlarged kidneys and hypodense lesions can be seen on CT scans. Other than IgG4-TIN, membranous nephropathy and mesangioproliferative glomerulonephritis may also develop in IgG4-related kidney disease, albeit this is unusual [10]. Membranous nephropathy can appear with nephrotic range proteinuria and hypoalbuminemia concomitant with TIN [10].
In those patients with elevated IgG4 levels, the severity of the disease and the serum IgG4 level frequently correlate. Serum and tissue IgG4 concentrations, however, do not serve as sensitive or specific indicators of IgG4-RD. A study revealed that serum IgG4 levels were normal in nearly half of active individuals with histologically confirmed IgG4-RD [11]. Particularly the retroperitoneum, involvement of specific organs or anatomical locations has a poorer connection with serum IgG4 levels. Consider false-negative IgG4 readings caused by the prozone phenomenon when there is multiorgan involvement and low blood IgG4 levels. Additionally, IgG4-RD frequently exhibits peripheral eosinophilia, high serum IgE levels, polyclonal hypergammaglobulinemia, high CRP, low titer positive ANA, rheumatoid factor, and hypocomplementemia. The majority of patients see a rapid drop in serum IgG4 levels after receiving glucocorticoids or B-cell depletion therapy, even if many of these patients do not return to normal levels when in clinical remission. Higher baseline levels of serum IgG4, IgE, and blood eosinophil concentrations in a prospective study of rituximab in IgG4-RD indicated greater risk of IgG4-RD recurrence and shorter time to relapse, making monitoring of these values important [12].
It is possible for IgG4-RD to advance from an inflammatory and proliferative stage that responds to treatments to a fibrotic stage that responds only weakly to treatments, which can result in serious organ damage. Because organ damage is permanent, prompt identification and therapy are crucial. Treatment goals include reducing side effects from glucocorticoids and other medications, causing the disease to remit, and maintaining organ function. Following diagnosis, a pretreatment evaluation should be conducted to gauge the extent and severity of the condition. Regular laboratory tests may be requested, including whole blood counts, kidney and liver function assessments, IgG subtype levels, IgE concentrations, serum C3 and C4 concentrations, urinalysis (asymptomatic proteinuria may be an indicator of TIN), chest, abdominal, and pelvic CT or magnetic resonance imaging (MRI) scans, and positron emission tomography (PET) scans (to assess the extent of the disease) [13].
Unless they are contraindicated, glucocorticoids are the primary line of treatment for patients who are treatment naive and want to induce remission. Prednisone monotherapy at a starting dose of 0.6 mg/kg/day (usually 30 - 40 mg/day) is advised. Within 2 - 4 weeks, almost all patients respond to 40 mg of prednisone daily. Many patients respond right away. Prednisone dose may be gradually decreased during 3 to 6 months till it is entirely withdrawn when clinical response is seen in the affected organ [14].
Remission maintenance therapy should be considered in a subset of high-risk patients, such as those with multiple organ involvement, high serum IgG4, IgE, and eosinophilia at the outset, as well as those with a history of relapse [15]. Recurrence was seen more commonly in patients identified at younger ages, those with allergy histories, and those whose therapy was started long after the diagnosis. In a recent retrospective study involving 277 IgG4-RD patients, recurrence was seen more commonly in patients identified at younger ages, those with allergy histories, and those whose therapy was started long after the diagnosis. At 12, 24, and 36 months, the cumulative relapse rates were 12.86%, 27.84%, and 36.1%, respectively. Regarding the organs affected by recurrence (125 organs, 101 patients), recurrence occurred in 40 patients in de novo organs and in 85 patients in the same organ. The parathyroid gland experienced the most frequent de novo recurrence. The thyroid gland, pancreas, and lacrimal gland were the three organs where recurrence in the same organ occurred most frequently [16].
Conclusions
This paper described a case of IgG4-RD involving the kidneys, in a patient with previous atopic dermatitis and recent development of unexplained pancreatitis and uveitis. Usually, IgG4-TIN is diagnosed in patients with known IgG4-RD elsewhere. In contrast, IgG4-TIN was the key to diagnosing IgG4-RD in our patient. It is also important to note that, in spite of initially negative serum IgG4-levels, the diagnosis still needs to be considered especially if multisystem involvement is present (as in this case). The patient was treated successfully with steroids and will be monitored clinically and biochemically for relapse or other organ involvement of IgG4-RD.
Learning points
The main “take-home” message of this case is the necessity of having heightened index of suspicion for IgG4-RD in the setting of multiple presenting symptoms. Even mundane history details, such as the presence of atopic dermatitis, may aid in including IgG4-RD on the differentials list. IgG4-RD of the kidneys may be the presentation which clinches the diagnosis.
Acknowledgments
None to declare.
Financial Disclosure
None to disclose.
Conflict of Interest
None to disclose.
Informed Consent
Verbal consent was obtained to use the case details in both written and visual media.
Author Contributions
All authors contributed to the writing/editing part of this paper. Mostafa Mohrag and Mohammed Abdulrasak wrote the manuscript. Mohammed Binsalman and Majid Darraj edited and guided us in writing this manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Lang D, Zwerina J, Pieringer H. IgG4-related disease: current challenges and future prospects. Ther Clin Risk Manag. 2016;12:189-199.
doi pubmed pmc - Okazaki K, Uchida K. Current perspectives on autoimmune pancreatitis and IgG4-related disease. Proc Jpn Acad Ser B Phys Biol Sci. 2018;94(10):412-427.
doi pubmed pmc - Islam AD, Selmi C, Datta-Mitra A, Sonu R, Chen M, Gershwin ME, Raychaudhuri SP. The changing faces of IgG4-related disease: Clinical manifestations and pathogenesis. Autoimmun Rev. 2015;14(10):914-922.
doi pubmed - Della-Torre E, Lanzillotta M, Doglioni C. Immunology of IgG4-related disease. Clin Exp Immunol. 2015;181(2):191-206.
doi pubmed pmc - Kamisawa T, Funata N, Hayashi Y, Eishi Y, Koike M, Tsuruta K, Okamoto A, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol. 2003;38(10):982-984.
doi pubmed - Perugino CA, AlSalem SB, Mattoo H, Della-Torre E, Mahajan V, Ganesh G, Allard-Chamard H, et al. Identification of galectin-3 as an autoantigen in patients with IgG(4)-related disease. J Allergy Clin Immunol. 2019;143(2):736-745.e736.
doi pubmed pmc - Kawa S, Ota M, Yoshizawa K, Horiuchi A, Hamano H, Ochi Y, Nakayama K, et al. HLA DRB10405-DQB10401 haplotype is associated with autoimmune pancreatitis in the Japanese population. Gastroenterology. 2002;122(5):1264-1269.
doi pubmed - Zen Y, Nakanuma Y. IgG4-related disease: a cross-sectional study of 114 cases. Am J Surg Pathol. 2010;34(12):1812-1819.
doi pubmed - Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, Kloppel G, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25(9):1181-1192.
doi pubmed - Alexander MP, Larsen CP, Gibson IW, Nasr SH, Sethi S, Fidler ME, Raissian Y, et al. Membranous glomerulonephritis is a manifestation of IgG4-related disease. Kidney Int. 2013;83(3):455-462.
doi pubmed - Wallace ZS, Deshpande V, Mattoo H, Mahajan VS, Kulikova M, Pillai S, Stone JH. IgG4-related disease: clinical and laboratory features in one hundred twenty-five patients. Arthritis Rheumatol. 2015;67(9):2466-2475.
doi pubmed pmc - Wallace ZS, Mattoo H, Mahajan VS, Kulikova M, Lu L, Deshpande V, Choi HK, et al. Predictors of disease relapse in IgG4-related disease following rituximab. Rheumatology (Oxford). 2016;55(6):1000-1008.
doi pubmed pmc - Stone JH, Patel VI, Oliveira GR, Stone JR. Case records of the Massachusetts General Hospital. Case 38-2012. A 60-year-old man with abdominal pain and aortic aneurysms. N Engl J Med. 2012;367(24):2335-2346.
doi pubmed - Carruthers MN, Topazian MD, Khosroshahi A, Witzig TE, Wallace ZS, Hart PA, Deshpande V, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis. 2015;74(6):1171-1177.
doi pubmed - Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN, Chari ST, et al. Second International Symposium on IgG4-Related Disease. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheumatol. 2015;67(7):1688-1699.
- Liu Y, Zeng Q, Zhu L, Gao J, Wang Z, Wang Z, Yang F, et al. Relapse predictors and serologically unstable condition of IgG4-related disease: a large Chinese cohort. Rheumatology (Oxford). 2020;59(8):2115-2123.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.