

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 15, Number 1, January 2024, pages 20-25

Hip Dysplasia in a Patient in Late Adolescence With Charcot-Marie-Tooth and Multiple Acyl-CoA Dehydrogenase Deficiency
Amirmohammad Heidaria, b, c, Cameron Stephena, d, Benan Dala-Alie, Jane Webbere, Oliver Pearcee, Mohamed H. Ahmedf, g, h, i
aFaculty of Medicine and Health Sciences, The University of Buckingham, Buckinghamshire, UK
bAintree University Hospital, Liverpool, UK
cUniversity of Liverpool, School of Medicine, Liverpool, UK
dAberdeen Royal Infirmary, Aberdeen, UK
eDepartment of Trauma and Orthopaedics, Milton Keynes Hospital NHS Foundation Trust, Milton Keynes, UK
fDepartment of Medicine and HIV Metabolic Clinic, Milton Keynes Hospital NHS Foundation Trust, Milton Keynes, UK
gDepartment of Geriatric Medicine, Milton Keynes Hospital NHS Foundation Trust, Milton Keynes, UK
hHonorary Senior Lecturer of the Faculty of Medicine and Health Sciences, University of Buckingham, UK
iCorresponding Author: Mohamed Hassan Ahmed, Department of Medicine and HIV Metabolic Clinic, Milton Keynes University Hospital NHS Foundation Trust, Eaglestone, Milton Keynes, Buckinghamshire, UK
Manuscript submitted November 21, 2023, accepted January 17, 2024, published online January 28, 2024
Short title: Hip Dysplasia in a Patient With CMT and MADD
doi: https://doi.org/10.14740/jmc4174
| Abstract | ▴Top |
This case report explores a unique presentation of hip dysplasia in a female patient aged 21 years old diagnosed with Charcot-Marie-Tooth disease (CMT) type 1A and multiple acyl-CoA dehydrogenase deficiency (MADD). The coexistence of these neuromuscular and metabolic disorders in a patient with hip dysplasia provides an opportunity to investigate their potential interactions and impact on diagnosis, treatment, and prognosis. The patient underwent labral repair with shelf osteotomy and later a total hip replacement. This case highlights the need for further research to better understand the relationships between CMT, MADD, neuromuscular dysplasia, and hip dysplasia. A deeper understanding of these interactions may lead to improved diagnostic techniques, earlier intervention, and personalized treatment approaches for patients with co-morbid conditions, ultimately improving patient outcomes and reducing complications later in life.
Keywords: Charcot-Marie-Tooth; Multiple acyl-CoA dehydrogenase deficiency; Hip prosthesis implantation; Orthopedics; Hip dysplasia
| Introduction | ▴Top |
Hip dysplasia is a relatively common orthopedic condition involving a spectrum of disorders affecting the hip joint in infants and children. Hip dysplasia can lead to high morbidity and decreased functional status [1]. This condition may coexist with various other syndromes, such as teratologic hip dysplasia and neuromuscular hip dysplasia, which can be frequently encountered in clinical practice.
Charcot-Marie-Tooth disease (CMT) and multiple acyl-CoA dehydrogenase deficiency (MADD) are neuromuscular and metabolic disorders, respectively. CMT or hereditary motor and sensory neuropathy is the most common inherited neuromuscular disorder. It is characterized by abnormal development of the peripheral nerves leading to muscle weakness that may impact hip joint stability [2]. It often presents with a pes cavus deformity (high-arched feet) in the adolescent years presenting numbness in the lower extremities, potentially leading to unintentional injury/ulceration, akin to that seen in diabetic peripheral neuropathy.
MADD is a rare autosomal recessive metabolic myopathy, characterized by impaired dehydrogenation reactions due to defective electron transfer from flavoprotein dehydrogenases to the mitochondrial respiratory chain. This results in a variety of consequences, which may present as: muscle weakness and poor exercise tolerance for age, lethargy, hypoglycemia, metabolic acidosis, and hepatomegaly [3].
Predominantly affecting cellular respiration and production of cellular energy resources, impacting tissues with high energy requirements, such as muscles, the coexistence of these conditions in a patient with hip dysplasia presents a unique opportunity to explore their potential interactions and their impact on the patient’s diagnosis, treatment, and prognosis. This case report illustrates a unique presentation of hip dysplasia in a female patient aged 21 years old diagnosed with CMT type 1A and MADD.
| Case Report | ▴Top |
Investigations
A female in her late adolescence presented to the emergency department with a 2-month history of right hip pain. The pain was specifically around the anterior and posterior aspects of the groin region. Her past medical history included CMT type 1A, MADD, asthma, adrenal insufficiency, inherited neuropathy, idiopathic toe walking, pes cavus and hammer toes. The patient had a background of multiple admissions with gastroenteritis and pneumonia. A diagnosis of MADD was suspected due to the family history. Late-onset MADD (type III) is the most common presentation, with signs and symptoms appearing anytime from infancy to adulthood. The patient’s genetic analysis for the electron transfer flavoprotein dehydrogenase (ETFDH) gene, associated with riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency, revealed the presence of a single mutation, which indicated of a carrier status. However, an abnormal plasma acylcarnitine profile, reflecting aberrant myoadenylate deaminase (MAD) enzyme activity, had been observed during a period of illness. It was hypothesized that the ’patient’s genetic alteration might have impacted their biochemical profile under specific stressors, such as infections, while remaining normal under other circumstances.
Her family history also indicated the presence of CMT neuropathy in multiple relatives, and the inheritance pattern might instead represent autosomal dominant inheritance with reduced penetrance. Additionally, the patient’s paternal family had a history of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency, a subtype of MADD.
The patient’s drug history included riboflavin (vitamin B2), morphine, codeine, zopiclone, omeprazole, cetirizine, prochlorperazine, beclomethasone and salbutamol inhalers, amitriptyline and tramadol.
The patient’s childhood and development history included being delivered via cesarean section with a breech presentation. Maternal history also revealed uterus didelphys. An ultrasound scan of both hips performed shortly after birth was reportedly normal. Although, early in her childhood, the patient had problems walking and received orthotic appliance treatment, serial ankle casts and botulinum toxin injections. Strong family history of CMT is also noted. She lives with parents and has never smoked and does not drink alcohol.
Diagnosis
On clinical examination, the patient had a thin build with symmetrical musculature in her lower limbs. She had an antalgic gait with normal lower limb alignment and no leg length discrepancy. Her right hip had a good range of movement with pain in extreme abduction and internal rotation.
A plain radiograph taken during her initial visit revealed evidence of right hip dysplasia with Shenton’s line intact and uncovering of the lateral side of the femoral head with minimal degenerative changes as illustrated in Figure 1.
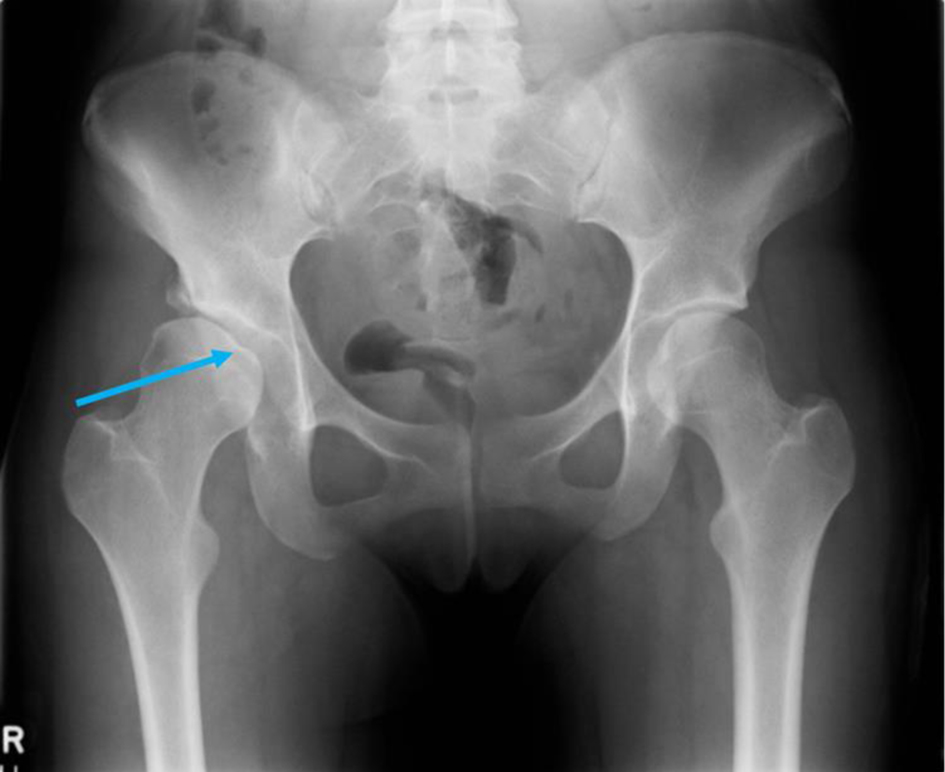 Click for large image | Figure 1. A plain radiograph taken during the first visit to the emergency department (ED), revealing right hip dysplasia, with uncovering of the lateral side of the femoral head and early secondary degenerative changes (blue arrow), and a normal appearance to the left hip on radiograph. |
The patient initially underwent a shelf osteotomy at the age of 18 years old (Fig. 2), to provide superior and supero-lateral femoral head coverage, and labral repair. The patient had a good result and was pain-free for 2 years, but the pain progressively returned. Subsequently, a magnetic resonance imaging (MRI) arthrogram of the right hip was performed (Fig. 3), which demonstrated a dysplastic hip with extensive abnormality of the acetabular rim and features of associated acetabular cartilage damage. The images were discussed at a multi-disciplinary meeting, and it was decided that symptomatically and radiologically a total hip replacement was indicated to address her pain and functional limitation, which was performed at the age of 21 years old (Fig. 4).
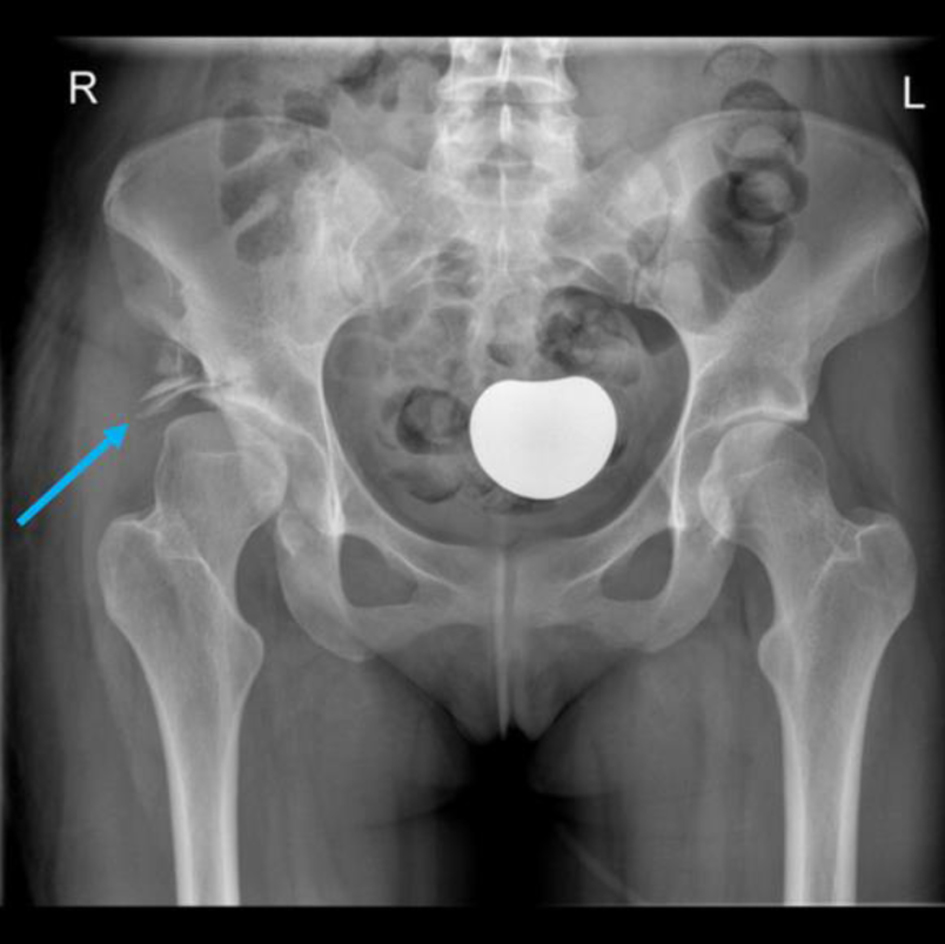 Click for large image | Figure 2. A plain radiograph after right labral repair with shelf osteotomy (blue arrow). |
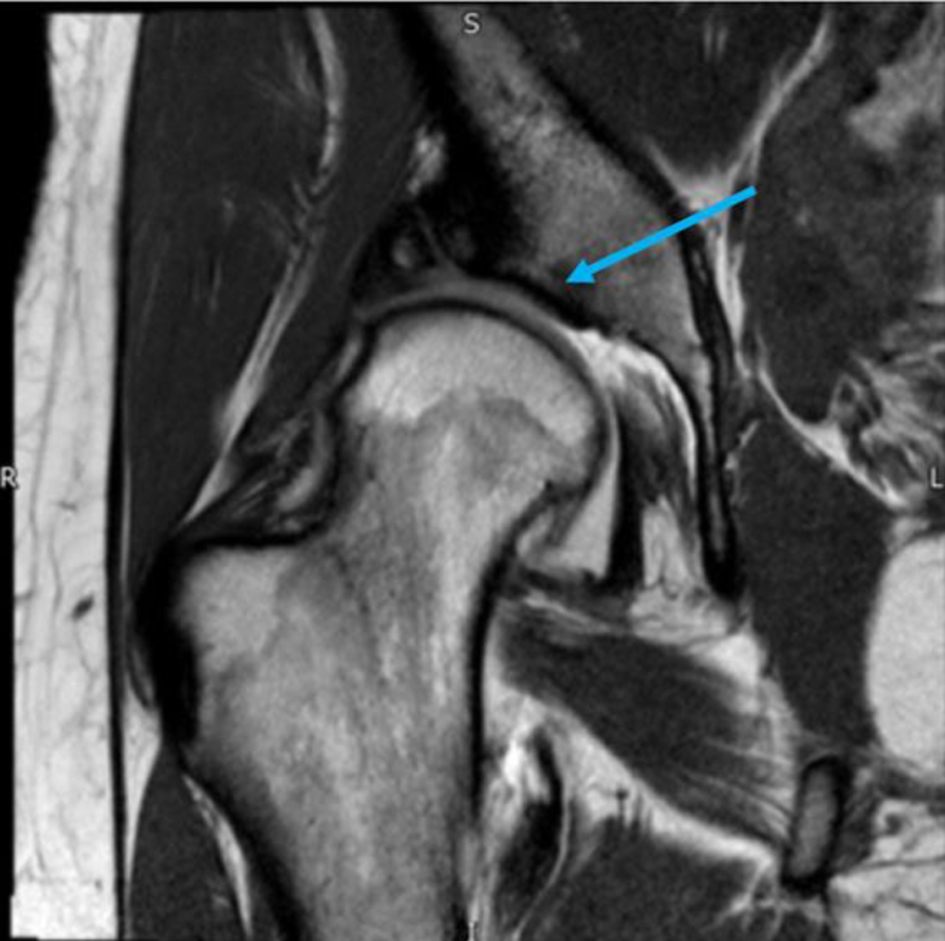 Click for large image | Figure 3. A magnetic resonance imaging (MRI) arthrogram of the right hip revealing a dysplastic hip and features of associated acetabular cartilage damage (blue arrow). |
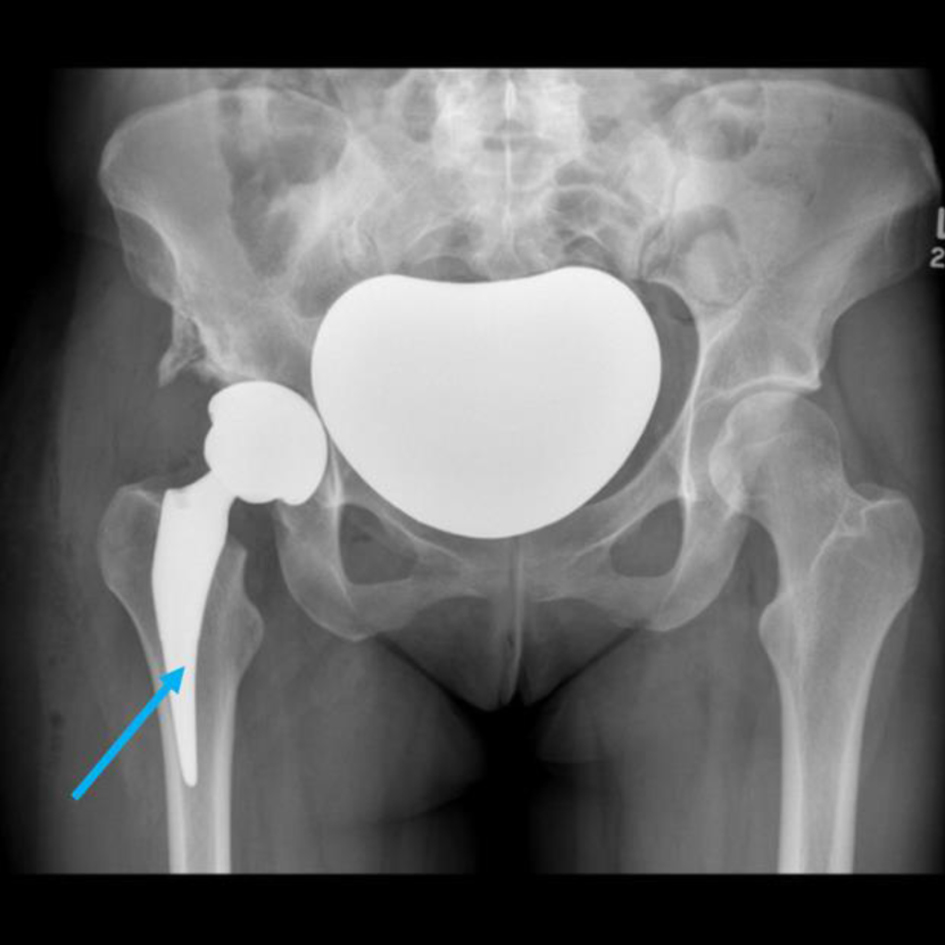 Click for large image | Figure 4. A plain radiograph showing a right total hip replacement (blue arrow). |
Follow-up and treatment
The patient has had several flare-ups of MADD and remains under ongoing monitoring by the endocrinology department at Oxford, with thorough evaluations being conducted at regular intervals. Her case has been referred to multiple multidisciplinary teams (MDTs) for psychological support and pain management, with a particular focus on neurology.
The patient is also currently under biannual surveillance by the chronic pain team at a tertiary center. Plans are being considered for potential discharge to community care, given stable and satisfactory responses to pain management interventions.
The patient has been experiencing challenges with dietary intake and is leaning more towards simple foods. They have been provided with nutritional supplements to help ensure proper nutrition. Additional support is being given by dietitian from a local tertiary team. The patient has been provided comprehensive follow-up care, with a keen focus on their overall health, return to daily activities, and the necessary aftercare arrangements.
| Discussion | ▴Top |
Hip dysplasia manifests itself in various types distinguished by etiology. Among these classifications are developmental dysplasia of the hip (DDH), neuromuscular developmental hip dysplasia (NMDH) and dysplasia secondary to metabolic or genetic disorders [4].
The terms DDH and NMDH are often used interchangeably. However, their exact relationship remains unclear. Some studies propose shared pathophysiological mechanisms between DDH and NMDH, such as abnormal muscle tone and joint laxity [5, 6]. Conversely, other studies highlight them as distinct conditions [7, 8]. There is general agreement on the increased incidence and severity of hip dysplasia in those with neuromuscular pathologies. For instance, the incidence of hip disorders is much higher in children with neuromuscular disorders, varying from 8% to 82% [9-11] and that NMDH generally presents later than DDH [10]. Also, the treatment for NMDH is predominantly surgical with some cases requiring numerous operations due to abnormal muscle tone [12, 13].
Different pathophysiologies underpin the disorders associated with upper motor neuron (UMN) and lower motor neuron (LMN) causes of hip dysplasia. In UMN disorders, factors such as spasticity, restricted mobility and functional muscular asymmetry exacerbate abnormalities of the femoral head and acetabulum, resulting in subluxation, dislocation and coxa valga [14-16]. Importantly, the common factors leading to coxa valga in CMT and MADD, are proximal weakness, limited weight-bearing, and ligamentous laxity [17, 18].
Research has highlighted a relationship between CMT and hip dysplasia, with the term Charcot-Marie-Tooth hip dysplasia (CMTHD) often used to describe the coexistence of the two conditions. It has been suggested that CMTHD patients present later in life [19, 20] and greater overall disordered hip morphology compared to that of DDH [21]. Typically, the hips of CMTHD patients are normal at birth unless there is concomitant DDH [22]. Furthermore, CMT patients have been reported with decreased bone mineral density, exacerbating the risk and severity of hip dysplasia [23, 24].
On the other hand, the role of MADD in hip dysplasia remains ambiguous. Characterized by the impairment of flavoprotein enzymes vital for energy production. MADD can lead to muscle weakness and instability of the hip joint due to accumulation of toxic metabolites [25, 26]. Metabolic acidosis, another consequence of MADD, can decrease bone mineral density, heightening susceptibility to deformities and instability (Fig. 5). Therefore, it is possible to suggest that the combination of these two conditions may have led to the NMDH. Other minor contributing factors are female sex and breech presentation.
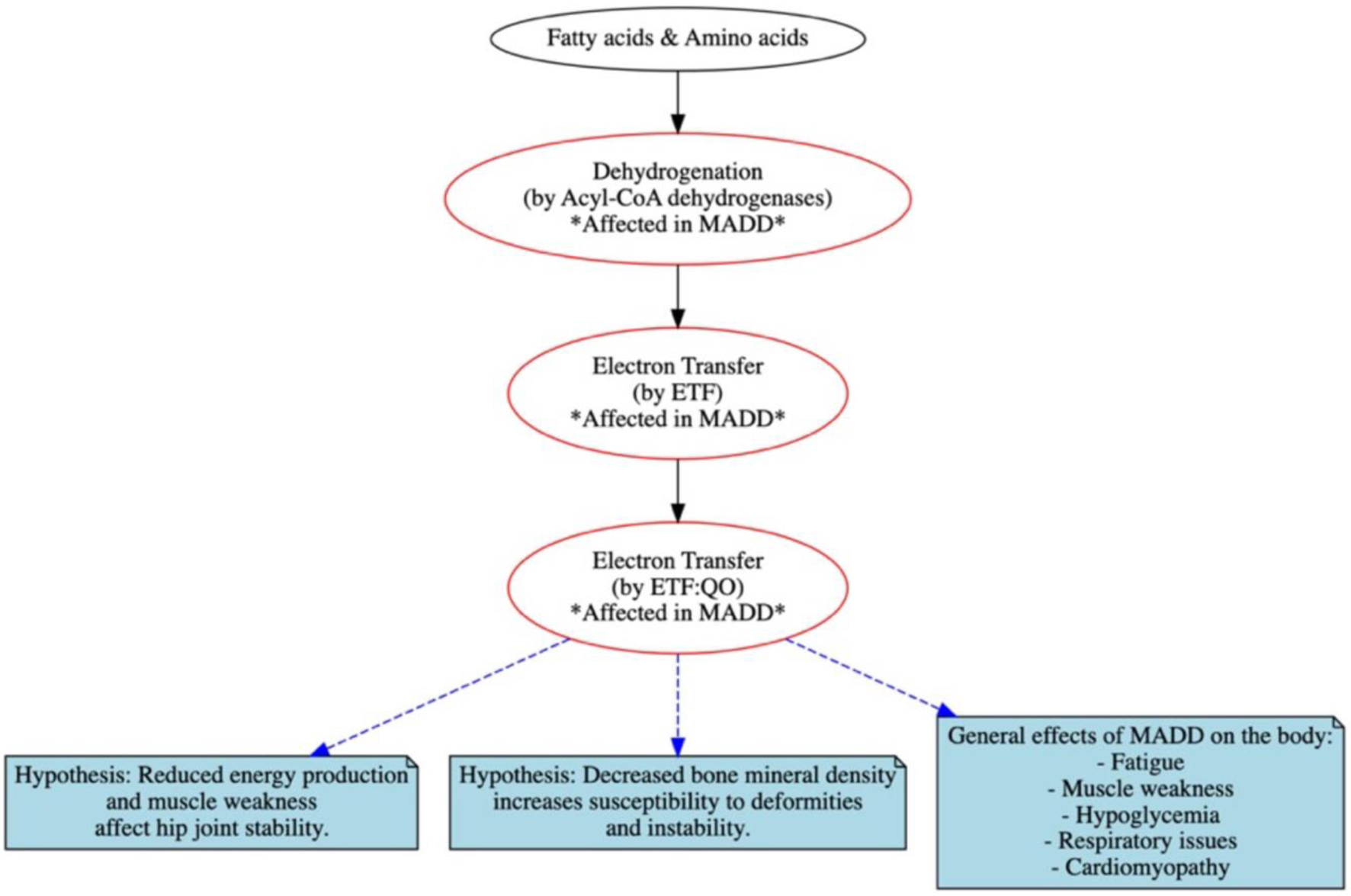 Click for large image | Figure 5. Schematic representation of key abnormalities including flavoprotein dehydrogenase enzymes, electron transfer flavoprotein (ETF) and ETF: ubiquinone oxidoreductase (ETF: QO) with outcomes in multiple acyl-CoA dehydrogenation deficiency (MADD). |
In analyzing this patient’s case, it is pertinent to consider potential confounding factors such as other genetic or environmental influences and the presence of other risk factors such as female sex and breech presentation. The varying severity and manifestation of CMT and MADD add complexity to understanding their impact on hip dysplasia. Born in breech presentation, the complexity is further compounded, highlighting the need for an evolving understanding of the mechanisms underlying CMT, MADD and hip dysplasia.
The unique interplay between NMDH, CMT, and MADD, as reflected in this patient’s case, underscores the intricacy of hip dysplasia. The presentation of ligamentous laxity, limited weight-bearing ability, proximal muscle weakness and decreased bone mineral density depicts the multifaceted nature of her condition. With MADD disrupting energy production, it could potentially exacerbate muscle weakness and hip joint instability. Coupled with neuromuscular issues stemming from CMT and NMDH, the MADD could contribute to the progression of hip dysplasia (Fig. 6).
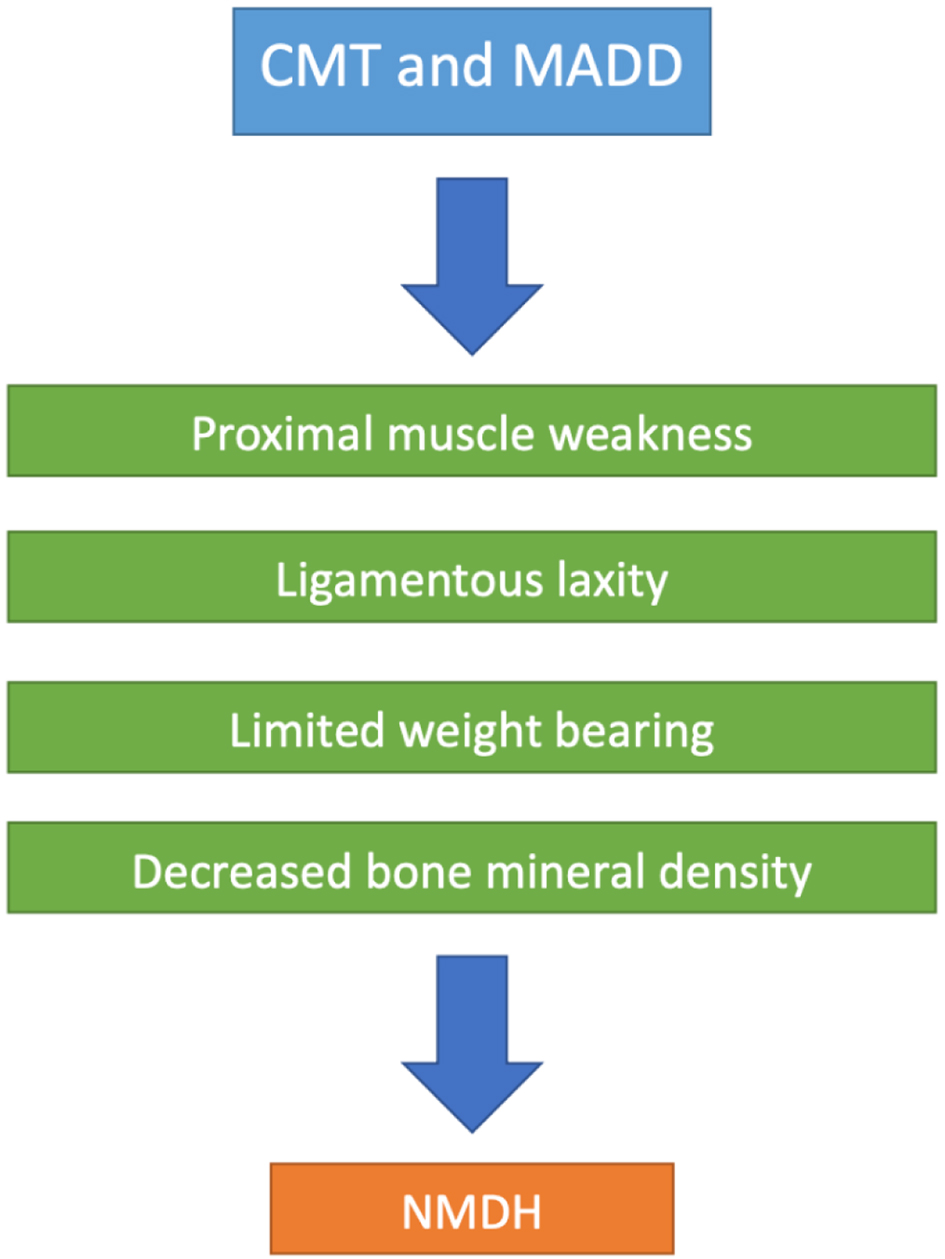 Click for large image | Figure 6. Suggested schematic representation of key abnormalities shared by Charcot-Marie-Tooth disease (CMT) and multiple acyl-CoA dehydrogenation deficiency (MADD) that in part can contribute to NMDH. |
Conclusions
This case reflects the complexity of hip dysplasia, particularly when coexisting with neuromuscular and metabolic disorders, emphasizing the need for ongoing research to gain a greater understanding. Developing more targeted therapeutic strategies, improving diagnostic techniques, and enabling earlier intervention are crucial to improving the quality of life for patients with similar co-morbid conditions. Identifying specific risk factors or biomarkers may aid in early detection, allowing for timely intervention and potentially delaying, or preventing, invasive procedures such as total hip replacements, ultimately leading to better patient outcomes with fewer complications in later life.
Learning points
The coexistence of neuromuscular disorders like CMT and metabolic disorders like MADD can complicate the presentation, diagnosis, and treatment of conditions like hip dysplasia. Hence, physicians should consider these potential interactions when diagnosing and treating such patients.
For patients presenting with complex cases of hip dysplasia, a multidisciplinary approach is essential for comprehensive patient management. Engaging with experts from various fields, such as endocrinology, neurology, orthopedics, and psychology, can lead to a more precise diagnosis and effective treatment plan.
Timely intervention plays a critical role in managing hip dysplasia, especially in patients with concomitant conditions like CMT and MADD. In this case, the patient underwent labral repair with shelf osteotomy followed by a total hip replacement, highlighting the importance of a proactive approach to surgical intervention.
This case underscores the need for further research into the relationships between neuromuscular disorders, metabolic disorders, and conditions like hip dysplasia. Such understanding may pave the way for improved diagnostic techniques, personalized treatment approaches, and ultimately better patient outcomes.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Informed consent has been obtained from the patient.
Author Contributions
Conception and design: MHA. Administrative support: all authors. Provision of study materials or patients: all authors. Collection and assembly of data: MHA, CA, and AH. Data analysis and interpretations: all authors. Manuscript writing: all authors. Final approval of manuscript: all authors.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Nandhagopal T, De Cicco FL. Developmental dysplasia of the hip. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2023.
pubmed pmc - Casasnovas C, Cano LM, Alberti A, Cespedes M, Rigo G. Charcot-Marie-Tooth disease. Foot Ankle Spec. 2008;1(6):350-354.
doi pubmed - Prasun P. Multiple acyl-CoA dehydrogenase deficiency. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al, editors. GeneReviews((R)). Seattle (WA): University of Washington; 1993.
pubmed pmc - Kraeutler MJ, Garabekyan T, Pascual-Garrido C, Mei-Dan O. Hip instability: a review of hip dysplasia and other contributing factors. Muscles Ligaments Tendons J. 2016;6(3):343-353.
doi pubmed pmc - Wilkinson J. Congenital displacement of the hip joint. London: Springer; 1985.
- Wilkinson JA, Sedgwick EM. Occult spinal dysraphism in established congenital dislocation of the hip. J Bone Joint Surg Br. 1988;70(5):744-749.
doi pubmed - Cooke PH, Cole WG, Carey RP. Dislocation of the hip in cerebral palsy. Natural history and predictability. J Bone Joint Surg Br. 1989;71(3):441-446.
doi pubmed - Luther AZ, Clarke NM. Developmental dysplasia of the hip and occult neurologic disorders. Clin Orthop Relat Res. 2008;466(4):871-877.
doi pubmed pmc - McCarthy JJ, Chafetz RS, Betz RR, Gaughan J. Incidence and degree of hip subluxation/dislocation in children with spinal cord injury. J Spinal Cord Med. 2004;27(Suppl 1):S80-83.
doi pubmed - Rink P, Miller F. Hip instability in spinal cord injury patients. J Pediatr Orthop. 1990;10(5):583-587.
doi pubmed - Vogel LC, Lubicky JP. Ambulation in children and adolescents with spinal cord injuries. J Pediatr Orthop. 1995;15(4):510-516.
doi pubmed - Samilson RL, Tsou P, Aamoth G, Green WM. Dislocation and subluxation of the hip in cerebral palsy. Pathogenesis, natural history and management. J Bone Joint Surg Am. 1972;54(4):863-873.
pubmed - Rueda J, Carroll NC. Hip instability in patients with myelomeningocele. J Bone Joint Surg Br. 1972;54(3):422-431.
pubmed - Spiegel DA, Flynn JM. Evaluation and treatment of hip dysplasia in cerebral palsy. Orthop Clin North Am. 2006;37(2):185-196.
doi pubmed - Moreau M, Drummond DS, Rogala E, Ashworth A, Porter T. Natural history of the dislocated hip in spastic cerebral palsy. Dev Med Child Neurol. 1979;21(6):749-753.
doi pubmed - Gamble JG, Rinsky LA, Bleck EE. Established hip dislocations in children with cerebral palsy. Clin Orthop Relat Res. 1990(253):90-99.
pubmed - Sporer SM, Smith BG. Hip dislocation in patients with spinal muscular atrophy. J Pediatr Orthop. 2003;23(1):10-14.
pubmed - Zenios M, Sampath J, Cole C, Khan T, Galasko CS. Operative treatment for hip subluxation in spinal muscular atrophy. J Bone Joint Surg Br. 2005;87(11):1541-1544.
doi pubmed - Coleman SS. Developmental dislocation of the hip: evolutionary changes in diagnosis and treatment. J Pediatr Orthop. 1994;14(1):1-2.
doi pubmed - Bamford NS, White KK, Robinett SA, Otto RK, Gospe SM, Jr. Neuromuscular hip dysplasia in Charcot-Marie-Tooth disease type 1A. Dev Med Child Neurol. 2009;51(5):408-411.
doi pubmed - Pouwels S, de Boer A, Leufkens HG, Weber WE, Cooper C, de Vries F. Risk of fracture in patients with Charcot-Marie-Tooth disease. Muscle Nerve. 2014;50(6):919-924.
doi pubmed - Abdala R, Levi L, Longobardi V, Zanchetta MB. Severe bone microarchitecture deterioration in a family with hereditary neuropathy: evidence of the key role of the mechanostat. Osteoporos Int. 2020;31(12):2477-2480.
doi pubmed - Novais EN, Bixby SD, Rennick J, Carry PM, Kim YJ, Millis MB. Hip dysplasia is more severe in Charcot-Marie-Tooth disease than in developmental dysplasia of the hip. Clin Orthop Relat Res. 2014;472(2):665-673.
doi pubmed pmc - Chan G, Bowen JR, Kumar SJ. Evaluation and treatment of hip dysplasia in Charcot-Marie-Tooth disease. Orthop Clin North Am. 2006;37(2):203-209.
doi pubmed - Lupica A, Oteri R, Volta S, Ghezzi D, Drago SFA, Rodolico C, Musumeci O, et al. Diagnostic challenges in late onset multiple acyl-CoA dehydrogenase deficiency: clinical, morphological, and genetic aspects. Front Neurol. 2022;13:815523.
doi pubmed pmc - Olsen RK, Olpin SE, Andresen BS, Miedzybrodzka ZH, Pourfarzam M, Merinero B, Frerman FE, et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain. 2007;130(Pt 8):2045-2054.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.