

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website https://www.journalmc.org |
Case Report
Volume 15, Number 10, October 2024, pages 278-282

Catastrophic Antiphospholipid Syndrome in a Lupus Patient With Severe Recurrent Life-Threatening Clinical Manifestations
Abdulrahman Ali M. Khormia, f ![]() , Maged Ba Gunaidb, Mohammed Fayyadc, Mostafa Mohragd, Ali Abdullah AlAseerie
, Maged Ba Gunaidb, Mohammed Fayyadc, Mostafa Mohragd, Ali Abdullah AlAseerie
aInternal Medicine and Rheumatology, Prince Sattam University College of Medicine, Al Kharj, Saudi Arabia
bInternal Medicine, Family Care Hospital, Riyadh, Saudi Arabia
cInternal Medicine and Nephrology, Family Care Hospital, Riyadh, Saudi Arabia
dInternal Medicine and Nephrology, Department of Medicine, Faculty of Medicine, Jazan University, Jazan 4514, Saudi Arabia
eInternal Medicine Department, Prince Sattam University College of Medicine, Al Kharj, Saudi Arabia
fCorresponding Author: Abdulrahman Ali M. Khormi, Internal Medicine and Rheumatology, Prince Sattam University College of Medicine, Al Kharj, Saudi Arabia
Manuscript submitted May 24, 2024, accepted August 12, 2024, published online September 20, 2024
Short title: CAPS in a Lupus Patient
doi: https://doi.org/10.14740/jmc4255
| Abstract | ▴Top |
Catastrophic antiphospholipid syndrome (CAPS) is a rare, severe, and life-threatening form of antiphospholipid syndrome (APS). Early recognition and rapid treatment are of great importance to improve patient outcomes and decrease mortality. Herein, we present a case of lupus and APS with obstetric complications, recurrent thrombosis, and renal and hematological manifestations of APS which showed great response to the treatment.
Keywords: Lupus; Antiphospholipid; Thrombosis; Nephritis; Thrombocytopenia
| Introduction | ▴Top |
Systemic lupus erythematosus (SLE) is a chronic multisystem autoimmune disease with various clinical manifestations that may affect any organ including the skin, blood, joints, kidneys, lungs, gastrointestinal system, and the nervous system. Although SLE’s etiology remains unclear, it is assumed that it is multifactorial including different environmental (e.g., ultraviolet light, infections, drugs, etc.), genetic, and hormonal factors.
Positive antiphospholipid antibodies are included in the immunological domain of New 2019 European League Against Rheumatism (EULAR)/American College of Rheumatology (ACR) classification criteria for SLE classification criteria [1, 2]. Antiphospholipid syndrome (APS) is an autoimmune disorder that can occur as an isolated primary phenomenon or secondary with other autoimmune diseases like SLE. APS is typified by a hypercoagulable state that can present as arterial or venous thrombosis. It can manifest in females who have experienced repeated miscarriages or fetal loss in the past. The revised criteria for the diagnosis of the syndrome consider three laboratory tests: the functional assay lupus anticoagulant (LAC), anti-cardiolipin (ACL), and antibodies against β2-glycoprotein I (aβ2GPI) [3-5]. Two labs and six clinical domains made up the new 2023 ACR/EULAR APS classification criteria. Clinical domains include hematological manifestation (thrombocytopenia). Patients are considered to have APS if they receive at least three points each from the clinical and laboratory domains combined [6]. Anticoagulation and antiplatelet therapy are the primary treatments for APS. Aspirin and other antiplatelets are frequently used to prevent arterial events in a primary prophylactic clinical setting, while anticoagulation is used for secondary prevention of venous or arterial thrombosis or as prophylaxis in high-risk patients [7].
Catastrophic antiphospholipid syndrome (CAPS) is an unusual, life-threatening form of APS. Herein, we report a case of lupus and CAPS presenting with obstetric complications, recurrent thrombosis, and renal and hematological manifestations of APS.
| Case Report | ▴Top |
Investigations
A 38-year-old pregnant woman (18 weeks) was referred to our hospital with severe generalized abdominal pain for 1 week duration. She was known to have lupus and positive APS antibodies for 5 years. She was only using on demand low dose of prednisone and aspirin. The patient was in her usual state of health and was on enoxaparin during her pregnancy. Abdominal pain developed suddenly and was associated with dyspnea, spotting, and undocumented fever. She was evaluated clinically by an obstetrician and by radiology ultrasound (US) and found to have intrauterine fetal death and possible liver infarction versus abscess. She underwent dilation and curettage (D&C), was admitted to critical care, and started on supportive care and antibiotic treatment. Our team was consulted to evaluate her from a rheumatological point of view, and she was seen within the same day of presentation. The patient gave a history of previous possible provoked deep vein thrombosis (DVT) and recurrent abortions. She had a history of chronic thrombocytopenia and anemia. Upon critical care admission, laboratory investigations revealed thrombocytopenia, anemia (hemoglobin (Hb): 8.4 g/dL, normal: 11 - 16), leukocytosis, elevated liver enzymes, low albumin, and significant proteinuria with preserved kidney function tests (KFTs), and disseminated intravascular coagulation (DIC) was suspected. Computed tomography (CT) scans of chest and abdomen were done and showed minimal bilateral pleural effusion, atelectasis but no evidence of pulmonary embolism (PE), hepatosplenomegaly with hypodense liver lesion (infarction), no evidence of mesenteric ischemia and no evidence of remaining fetal products (Fig. 1). The patient was covered with antibiotic treatment, prophylactic heparin and steroid treatment. A few days later, she developed sudden new onset acute chest pain. Electrocardiogram (ECG) showed no acute ischemic changes, but she had elevated cardiac enzymes and high pro B-type natriuretic peptide (proBNP). A transthoracic echocardiogram (ECHO) was arranged and showed normal ejection fraction (EF) with moderate to severe mitral regurgitation. Other available lab results showed normal KFT, urine dipstick: +4 blood, +2 protein, red blood cells (RBCs) of over 100, white blood cells of 5 - 10, protein/creatinine (Cr) ratio of 1,528 mg/g (abnormal), C-reactive protein (CRP) of 546 mg/L (high), and erythrocyte sedimentation rate (ESR) of 130 mm/h (high). Blood smear and morphology showed no fragmented cells, lactate dehydrogenase (LDH) of 887 U/L (high), fibrin degradation products (FDPs) > 20, fibrinogen of 5.4 (high), normal total and direct bilirubin, antinuclear antibody (ANA) of 1:640, C3 of 90.04 mg/dL (normal), and C4 of 8.05 mg/dL (low). Heparin-induced thrombocytopenia (HIT) study was negative, anti-beta-2-glycoprotin IgG was 14.2 RU/mL (normal), IgM was 38.3 RU/mL (high), ACL ab IgG was 7.2 U/mL (negative), IgM was 17 U/mL (low positive), LAC was 115 (high), perinuclear antineutrophil cytoplasmic antibody (p-ANCA), cytoplasmic ANCA (c-ANCA) and anti-glomerular basement membrane (anti-GBM) were negative, hepatitis screen was negative, human immunodeficiency virus (HIV) was negative, D-dimer was 8.9 U/mL (high), and N-terminal (NT)-proBNP was 4,188 pg/mL (high). Computed tomography pulmonary angiogram (CTPE) study confirmed new subsegmental PE and kidney biopsy was arranged later and revealed thrombotic microangiopathy (TMA) consistent with APS (Fig. 2).
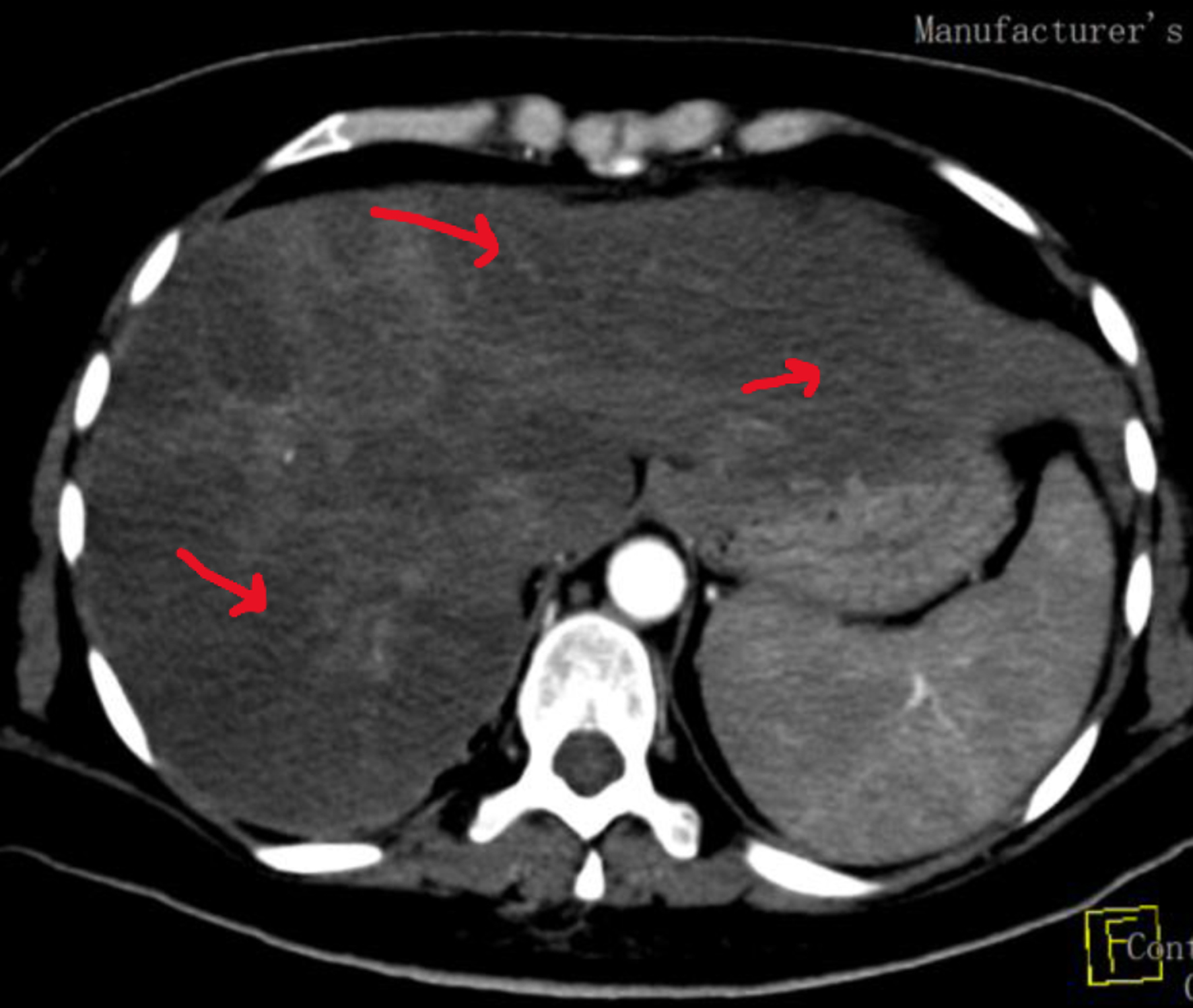 Click for large image | Figure 1. Computed tomography image of abdomen upon diagnosis. Arrows show multiple focal hypoattenuation lesions mainly at right hepatic lobe and irregular margins with marginal enhancement lesion and multiple hepatic infarcts. |
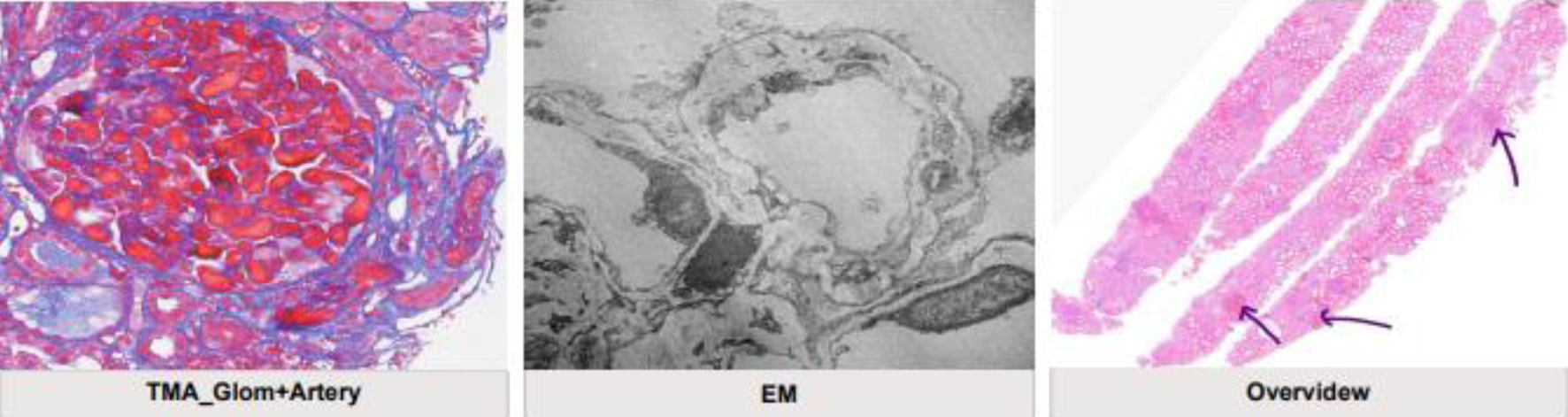 Click for large image | Figure 2. Kidney biopsy results showing extensive thrombotic microangiopathy consistent with antiphospholipid syndrome. Extensive glomerular and focal arterial recent thrombotic microangiopathy is seen with ischemic changes of perfused glomeruli. Immunofluorescence microscopy and electron microscopy have not shown any evidence of immune complex deposits therefore a concomitant lupus nephritis is unlikely. Electron microscopy shows re-modeling of glomerular basement membrane consistent and significant podocytopathy. Mild chronic parenchymatous damage is seen (arrows). |
Treatment
Providing the clinical manifestation, laboratory findings, renal biopsy results and images which instituted the multi-organ involvement (heart, lungs, liver, kidneys), a CAPS diagnosis was raised. Pulse steroid treatment was commenced, and the patient was referred for plasma exchange beside anticoagulation treatment. A significant improvement in her general condition and laboratory modalities was observed and the patient was discharged on tapering steroid dose and anticoagulation treatment. Hydroxychloroquine 400 mg daily was started, and a close follow-up appointment was given. Before discharge, laboratory results showed Hb from 8.4 to 10.9, platelets from 48 to 128, protein/creatinine ratio from 1,528 to 75, partial thromboplastin time (PTT) from 50.6 to 28.7, CRP from 546 to 29.6, and ESR from 130 to 40.
Follow-up
The patient has been undergoing regular follow-up with Nephrology and Rheumatology teams to taper down the patient’s steroid with regular check of anti-coagulation profile. Upon a follow-up visit, patient experienced right upper quadrant abdominal pain and was referred to gastroenterology team and after multiple investigations including US of abdomen, magnetic resonance cholangiopancreatography (MRCP) and full clinical laboratory evaluation, a cholecystitis diagnosis was confirmed. Patient was covered with antibiotic treatment and managed conservatively and improved significantly. Later, the patient experienced significant proximal lower limbs weakness with preserved reflexes and sensory exam. Steroid and hydroxychloroquine-induced myopathy were suspected. Hydroxychloroquine was stopped and steroid tapered to low dose. The myopathy improved significantly, and the patient returned to her normal life. She will keep following up with rheumatology, hematology, and nephrology teams.
| Discussion | ▴Top |
Additional related signs of APS that emphasize its systemic nature are cytopenias, cognitive dysfunction, heart valve disease, renal impairment, and skin ulcers. Furthermore, at the diagnosis stage, consideration should be given to related disorders such as infection. About half of APS cases occur with SLE and the remaining cases are designated as “primary APS”. The most severe and uncommon form of APS is called CAPS. It is characterized by large- and microvascular-vessel thrombosis, numerous organ dysfunction, and severe thrombotic consequences that develop quickly. It affects roughly 1% of patients with APS and, if left untreated, has a significant death rate. Early identification and prompt treatment may increase survival [8-10].
APS is undoubtedly an autoimmune condition, yet there is currently no effective immunomodulatory therapy for it. For individuals with APS, vitamin K antagonists are the mainstay of care in preventing thrombotic problems that relapse. Treatments for all patients with CAPS should involve anticoagulation, corticosteroids, and plasma exchange, either in conjunction with IVIG or not. Cyclophosphamide should also be taken into consideration for SLE patients. Furthermore, alternative treatments like rituximab and eculizumab may be taken into consideration in cases of relapse [7, 10].
Cervera et al reviewed 97 patients with intestinal involvement secondary to the APS. Thirty-seven patients were diagnosed with classic APS and 60 with CAPS. It was observed that in patients with classic APS, the prevalence of abdominal pain as the initial sign of intestinal ischemia was higher (76% versus 37%; P < 0.005), but patients with CAPS had a higher death rate (55% versus 17%; P < 0.0005) [11].
Stammler et al reported two cases of females who were triple-positive for antiphospholipid antibodies. The first patient, with history of previous thrombotic event, had multiorgan failure 3 days after vitamin K antagonists were replaced by rivaroxaban, and the second developed a similar attack 7 days after introduction of the same treatment. Both cases are CAPS episodes and were treated effectively with heparin followed by vitamin K antagonists, corticosteroids, and plasmapheresis. These two cases prove the inefficacy of rivaroxaban [12].
This was a challenging case because of the uncertainty of initial diagnosis, SLE vs. sepsis and DIC, and the need for confirmation of her APS diagnosis. Patient at time of presentation with sequel of intrauterine fetal death, persistent refractory abdominal pain and multi hepatic lesions raised question of APS. Renal involvement was another challenge. Lupus nephritis and sepsis-induced acute kidney injury beside APS were top differential diagnoses. Luckily, renal biopsy confirmed TMA and APS.
Conclusion
CAPS is an unusual, life-threatening form of APS. Early recognition and rapid treatment are of great importance to improve patient outcomes. We present a case of lupus and APS with obstetric complications, recurrent thrombosis, and renal and hematological manifestations of APS which showed great response to the treatment.
The learning points from this case include: 1) the importance of early recognition and treatment of CAPS to improve outcome; 2) the crucial role of renal biopsy to differentiate between different causes of renal involvement (SLE vs. APS in our case) as the treatment of APS will require long-term anticoagulation and the two conditions carry different characters and treatment options; and 3) finally, the importance of close follow-up to assess patient response to the treatment and to monitor any medication side effects and to manage accordingly.
Acknowledgments
We extend our appreciation to Prince Sattam bin Abdulaziz University, Deanship of Scientific Research for their support of this work.
Financial Disclosure
This paper did not receive a grant from any sector.
Conflict of Interest
The authors declare no conflict of interest.
Informed Consent
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Author Contributions
AAK conducted literature review, provided writing materials, and commenced at writing initial and final draft of the article. MBG and MF collected and organized patient data. Also, they provided logistic support and helped write the paper. AAA and MM reviewed the initial draft, provided corrections, helped review the literature and finalized the paper. ALL authors have critically reviewed and approved the final draft and are responsible for the content and similarity index of the manuscript.
Data Availability
Any inquiries regarding supporting data availability of this study should be directed to the corresponding author upon reasonable request.
Abbreviations
ACL: anti-cardiolipin; ANA: antinuclear antibody; APS: antiphospholipid syndrome; BNP: brain natriuretic peptide; CAPS: catastrophic antiphospholipid syndrome; CBC: complete blood count; Cr: creatinine; CRP: C-reactive protein; CT: computed tomography; D&C: dilation and curettage; DIC: disseminated intravascular coagulation; ECG: electrocardiogram; ECHO: echocardiogram; EF: ejection fraction; ESR: erythrocyte sedimentation rate; KFT: kidney function test; LAC: lupus anticoagulant; MRCP: magnetic resonance cholangiopancreatography; PE: pulmonary embolism; SLE: systemic lupus erythematosus; TMA: thrombotic microangiopathy; US: ultrasound
| References | ▴Top |
- Lisnevskaia L, Murphy G, Isenberg D. Systemic lupus erythematosus. Lancet. 2014;384(9957):1878-1888.
doi pubmed - Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, Smolen JS, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Ann Rheum Dis. 2019;78(9):1151-1159.
doi pubmed - Khamashta MA, Bertolaccini ML, Hughes GR. Antiphospholipid (Hughes) syndrome. Autoimmunity. 2004;37(4):309-312.
doi pubmed - Atanassova PA. Antiphospholipid syndrome and vascular ischemic (occlusive) diseases: an overview. Yonsei Med J. 2007;48(6):901-926.
doi pubmed pmc - Mazzoccoli C, Comitangelo D, D'Introno A, Mastropierro V, Sabba C, Perrone A. Antiphospholipid syndrome: a case report with an unusual wide spectrum of clinical manifestations. Auto Immun Highlights. 2019;10(1):9.
doi pubmed pmc - Barbhaiya M, Zuily S, Naden R, Hendry A, Manneville F, Amigo MC, Amoura Z, et al. 2023 ACR/EULAR antiphospholipid syndrome classification criteria. Ann Rheum Dis. 2023;82(10):1258-1270.
doi pubmed - Abdulrahman ZA, Azeez H, Hassan R, Ng J, Kaell A. A case report of anti-phospholipid syndrome with lower extremity arterial thrombosis that didn't respond to heparin and direct oral anticoagulation: ultimately, the patient agreed to oral warfarin. Cureus. 2022;14(11):e31230.
doi pubmed pmc - Cervera R, Rodriguez-Pinto I, Legault K, Erkan D. 16th international congress on antiphospholipid antibodies task force report on catastrophic antiphospholipid syndrome. Lupus. 2020;29(12):1594-1600.
doi pubmed - Bucciarelli S, Espinosa G, Cervera R, Erkan D, Gomez-Puerta JA, Ramos-Casals M, Font J, et al. Mortality in the catastrophic antiphospholipid syndrome: causes of death and prognostic factors in a series of 250 patients. Arthritis Rheum. 2006;54(8):2568-2576.
doi pubmed - Kazzaz NM, McCune WJ, Knight JS. Treatment of catastrophic antiphospholipid syndrome. Curr Opin Rheumatol. 2016;28(3):218-227.
doi pubmed pmc - Cervera R, Espinosa G, Cordero A, Oltra MR, Unzurrunzaga A, Rossinol T, Plaza J, et al. Intestinal involvement secondary to the antiphospholipid syndrome (APS): clinical and immunologic characteristics of 97 patients: comparison of classic and catastrophic APS. Semin Arthritis Rheum. 2007;36(5):287-296.
doi pubmed - Stammler R, Legendre P, Cacoub P, Blanche P, Piette JC, Costedoat-Chalumeau N. Catastrophic antiphospholipid syndrome following the introduction of rivaroxaban. Lupus. 2020;29(7):787-790.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.