

| Journal of Medical Cases, ISSN 1923-4155 print, 1923-4163 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Med Cases and Elmer Press Inc |
| Journal website http://www.journalmc.org |
Case Report
Volume 3, Number 6, December 2012, pages 375-378
Unusual Presentation of a Dysembryoplastic Neuroepithelial Tumor Mimicking a Pontomesencephalic Cavernous Hemangioma
Daniela Hoehna, b, c, Sergej Konopleva, C. Cameron Yina
aColumbia University Medical Center, Department of Pathology and Cell Biology, Division of Hematopathology, New York, New York, 10032, USA
bDepartment of Hematopathology, UT MD Anderson Cancer Center, Houston, TX, USA
cCorresponding author: Daniela Hoehn, Department of Pathology and Cell Biology, 630 West 168th Street, VC14-239, Columbia University Medical Center & New York Presbyterian Hospital, New York, New York 10032, USA
Manuscript accepted for publication May 24, 2012
Short title: Dysembryoplastic Neuroepithelial Tumor
doi: https://doi.org/10.4021/jmc718w
| Abstract | ▴Top |
Dysembryoplastic neuroepithelial tumor is a rare benign neoplasm that usually occurs in children and young adults and is characterized by intractable seizures that are refractory to medication. Since its description and inclusion in the World Health Organization classification of brain tumors, an increasing number of cases have been reported. The tumor is preferentially, but not exclusively, located in the supratentorial cortex. Rare cases of other locations have been reported. We describe the clinical, imaging and pathologic features of a case of dysembryoplastic neuroepithelial tumor arising in the pons of a 26-year-old woman, an unusual extracortical location, mimicking a cavernous hemangioma.
Keywords: Dysembryoplastic neuroepithelial tumor; Pontomesencephalic; Cavernous hemangioma
| Introduction | ▴Top |
Dysembryoplastic neuroepithelial tumors (DNET) are rare benign neoplasms of the central nervous system that almost always manifest as medically intractable seizure in children and young adults. The term DNET was proposed after its identification in 1988, and has been used to encompass a spectrum of low-grade, mixed neuronal and glial neoplasms with little growth potential that may look like astrocytomas, oligodendrogliomas, or mixed gliomas [1]. Some variations of these tumors were described at the 2nd meeting of World Health Organization (WHO) in Zurich in 1990. This resulted in the incorporation of these tumors into the category of low-grade (WHO grade 1) neuronal and mixed neuronal glial tumors in the revised WHO classification in 1993 [2]. Dysembryoplastic neuroepithelial tumors most often arise in the temporal or frontal lobes, and are characterized by hypointensity on computerized comography (CT) scan, multinodular architecture and heterogeneous cellular composition. Other sites or origin are very rare [3-7]. Although most patients present with a history of long-standing seizures that are refractory to medication, rare presentations with clinically silent, chronic hemorrhagic depositions simulating a cavernous hemoangioma on magnetic resonance imaging (MRI) have been reported [8]. We report a unique case of DNET at the pontomesencephalic junction with acute spontaneous hemorrhage, which complicated the clinical presentation and blurred the differential diagnoses.
| Case Report | ▴Top |
The patient was a 26-year-old woman with no significant past medical history. She presented with a sudden onset of articulation disorder (slurred speech). She reported light headache, double vision and a perioral dysesthesia which rapidly progressed to a left sided hemidysesthesia. She also experienced, right sided loss of coordination and balance resulting in ataxia. All cognitive functions remained unimpaired. No syncopal events or epileptic episodes were described at any time. There was no history of vertigo or vomiting. An emergency contrast enhanced CT scan revealed a large (17 mm in diameter), well-demarcated, subcortical hyperdense lesion in the right mesencephalon with extension into the tegmentum pontis (Fig. 1A). No mass effect was identified. Ring-shaped hyperdensities and mild peritumoral edema were encompassed diagnostic of exuberant calcifications and secondary hemorrhagic infarction.
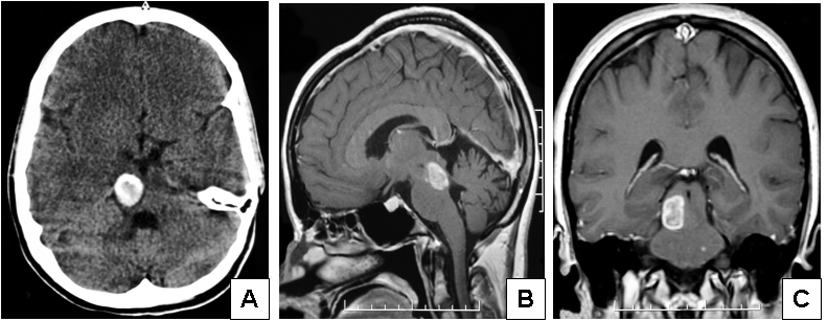 Click for large image | Figure 1. Imaging features of the case of dysembryoplastic neuroepithelial tumor presenting in the pontomesencephalic area. (A) Contrast enhanced CT scan showed a large, well-demarcated, subcortical hyperdense lesion in the right mesencephalon with extension into the tegmentum pontis. (B, lateral) and (C, coronal) MRI examination revealed a pontomesencephalic pseudocystic lesion that was hyperintense on T1 weighted images and showed less enhancement on T2 weighted images extending within the brain stem and reaching the red nucleus, the superior colliculi and the superior cerebellar tract. |
With the suspicion of an acute bleeding from a brainstem cavernoma, the patient was referred to the department of neurosurgery for evacuation. Preoperative laboratory values were unremarkable and non-contributory. MRI examination revealed a pontomesencephalic pseudocystic lesion that was hyperintense on T1 weighted images and showed less enhancement on T2 weighted images extending within the brain stem and reaching the red nucleus, the superior colliculi and the superior cerebellar tract (Fig. 1B, C). No mass effect, aqueductal obstruction, or intraventricular extension of the tumor was detected by either CT or MRI scans. Based on these findings, a diagnosis of cavernous angioma or cavernoma was rendered. The patient was placed on seizure prophylaxis, and was scheduled for surgical removal of the lesion.
A median occipito-suboccipital craniotomy was performed while the patient was in a sitting position. Intraoperative neurophysiological monitoring/cranial nerve monitoring was performed and the lesion was removed through a right trans-uvulo-tonisllar fissure via a fourth ventricle approach. The tissue around the lesion showed xanthochromic color. Instead of a cavernous angioma, a glial-like tumor with intratumoral calcifications was found. Spontaneous discharges in the facial nerve nucleus led to only a partial resection of the lesion and a small remnant of tumor was left in the caudal and lateral aspects in order to preserve facial nerve function. Histologic examination revealed a predominantly microcystic mucinous structure with randomly distributed mature appearing neurons and multiple psammoma bodies. A minor angiomatous component with hamartomatous vessels and capillaries was also identified, as usually seen in cavernomas. Hemosiderin deposition was demonstrated by Perl’s stain. In addition, some oligodendrocytic and glial elements were observed (Fig. 2). Based on these overall findings, a diagnosis of DNET was made.
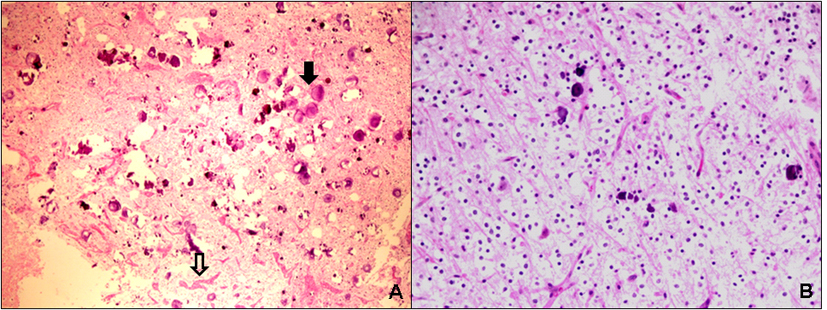 Click for large image | Figure 2. Microscopic features of the case of dysembryoplastic neuroepithelial tumour presenting in the pontomesencephalic area. (A) Low power view demonstrates a loosely structured mucinous background with numerous psammomatous calcifications (solid arrow) and areas of hamartomatous vasculature (open arrow) (H&E, x 100). (B) Medium power view reveals randomly distributed mature-appearing oligodendroglia-like cells suspended in a partially microcystic mucinous background (“floating-neurons”) (H&E, x 200). |
The patient did not manifest any new neurologic deficits postoperatively, and articulation, dysesthesia and gait ataxia improved during the following days.
| Discussion | ▴Top |
Dysembryoplastic neuroepithelial tumors are rare benign lesions that usually occur in patients younger than 20 years old and most often present as a long history of refractory seizures and cortical dysplasia. Other symptoms may include headache and nausea, as well as recognition, movement and sensation disorder, depending on the location of the lesion. The tumors have a very stable clinical course, and surgical resection without any adjuvant therapy remains the treatment of choice. As a result, accurate identification of DNET is critical in terms of rendering a timely diagnosis to initiate the appropriate treatment, avoiding exacerbation of seizures, and chemotherapy or radiation therapy interventions.
The characteristic temporal and frontal lobe location and a history of drug-resistant complex partial seizures, whenever present, remain the most important clinical features that can help to establish a diagnosis of DNET. Although originally thought to be restricted to the supratentorial cortex, DNET have been described in other locations [3-7]. Atypical presentations other than drug-resistant seizures have also been reported. Therefore it becomes a necessity to formulate the diagnosis as a cooperated multidisciplinary interaction among neurologists, neuroradiologists, neurosurgeons and neuropathologists. However, reports of unusual locations have been gradually expanding during the last decades, and include but not limit to brain stem [4], cerebellum [4], basal ganglia and thalamus [9]. It is established that these locations correspond to the topography of secondary germinal layers, the putative origin of DNET. In our patient, both aspects, unusal location and presentation complicated rendering the proper diagnosis. The tumor arose at the pontomesencephalic junction, and epileptic episodes were not noted at any time. Instead, there seemed to be a sudden onset in the development of neural deficits similar to symptoms of an acute haemorrhagic incident. The patient had no coagulopathy or any related bleeding disorders. While it is well recognized that primary intracranial neoplasms are often accompanied by prominent vascular abnormalities with resultant intratumorous hemorrhage, these episodes may mask their initial presentation [10].
Neuroimaging findings may serve as a very useful tool for initial evaluation and diagnosis of DNET [11]. Imaging characteristics of DNET are similar to those of other low-grade glial tumors. The tumors manifest as a hypoattenuated mass with or without calcification on CT. On MRI studies they present as cortical masses that are hypointense on T1-weighted images and hyperintense on T2-weighted images without surrounding vasogenic edema. To facilitate the proper identification of these lesions, three architectural features are frequently observed on MRI and could serve as a clue for the diagnosis of DNET [11]. First, the tumors are usually multinodular or multicystic. Second, the tumor width is often maximal at the cortical level and decreased toward the ventricles, leading to a triangular pattern of distribution best seen on coronal images. Third, most of the lesions seem to be divided by thin septa best seen on high-resolution MRI (“septations”). However, none of these features are solely characteristic and diagnostic for DNET, and whereas contrast enhancement and calcifications have been described [12], these seem to be rare occurrences. Our case further emphasized that the imaging appearance of DNET can be significantly altered and misleading, therefore complicating radiologic assessment and impeding the formulation of differential diagnoses.
Grossly, DNET may show a wide variation in size and shape, but are usually mucinous, multinodular or muticystic lesions of friable consistency [1]. Intraoperatively the tumors often exhibit a thickened cortex with a blurred border between the grey and white matter.
Microscopically, DNET are composed of multiple nodules that vary in size but all contain both neuronal and glial components. The cells are bland resembling mature oligodendrocytes, astrocytes and neurons that appear to “float” within mucin pools near bundles of axons flanked by oligodendroglia. Mitotic figures are rarely appreciated. Immunohistochemical stain can facilitate the identification of the glial component (GFAP), oligodendroglia-like cells (S-100), neurons (NeuN, synaptophysin) and neurofilaments (neuron specific enolase). The mucinous background can be highlighted with Alcian blue.
The primary differential diagnoses include oligodendrogliomas, mixed gliomas and gangliogliomas. The above-mentioned clinical, imaging and histologic features are the key in making the distinction. In our case, although the location of the tumor, the clinical presentation and the radiologic findings are atypical, the proper diagnosis could be established based on the histologic features, including a multinodular and multicystic gross appearance and the presence of both neuronal and glial components microscopically.
To the best of our knowledge, this is the first case of a calcified DNET arising at the pontomesencephalic junction. The clinical and neuroradiological presentation was falsely suggestive of a cavernoma with acute hemorrhage. Hence, we suggest to add dysembryoplastic neuroepithelial tumors to the list of differential diagnoses of lesions in this area and to take into consideration the risk of potential intratumoral bleeding when dealing with such neoplasm.
| References | ▴Top |
- Daumas-Duport C, Scheithauer BW, Chodkiewicz JP, Laws ER, Jr., Vedrenne C. Dysembryoplastic neuroepithelial tumor: a surgically curable tumor of young patients with intractable partial seizures. Report of thirty-nine cases. Neurosurgery. 1988;23(5):545-556.
pubmed doi - Daumas-Duport C, Pietsch T, Lantos PL. Dysembryoplastic neuroepithelial tumor. In: Kleihues P, Cavenee WK (eds): World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of the Nervous System. Lyon: IARC Press, 2000, pp103-106.
- Argyropoulou MI, Arvanitis DL, Tzoufi M, Fanis SL, Papadopoulou ZL, Efremidis SC. Dysembryoplastic neuroepithelial tumour and cerebellar atrophy: case report. Neuroradiology. 2001;43(1):73-75.
pubmed doi - Fujimoto K, Ohnishi H, Tsujimoto M, Hoshida T, Nakazato Y. Dysembryoplastic neuroepithelial tumor of the cerebellum and brainstem. Case report. J Neurosurg. 2000;93(3):487-489.
pubmed doi - Kurtkaya-Yapicier O, Elmaci I, Boran B, Kilic T, Sav A, Pamir MN. Dysembryoplastic neuroepithelial tumor of the midbrain tectum: a case report. Brain Tumor Pathol. 2002;19(2):97-100.
pubmed doi - Cervera-Pierot P, Varlet P, Chodkiewicz JP, Daumas-Duport C. Dysembryoplastic neuroepithelial tumors located in the caudate nucleus area: report of four cases. Neurosurgery. 1997;40(5):1065-1069; discussion 1069-1070.
pubmed doi - Arbol D, Gandotra P, Maqbool M, Shah A, Ahmad S. Dysembryoplastic neuroepithelial tumor: a rare brain tumor presenting with atypical radiological findings. JK Science 2007;9:145-147.
- Thom M, Gomez-Anson B, Revesz T, Harkness W, O'Brien CJ, Kett-White R, Jones EW, et al. Spontaneous intralesional haemorrhage in dysembryoplastic neuroepithelial tumours: a series of five cases. J Neurol Neurosurg Psychiatry. 1999;67(1):97-101.
pubmed doi - Leung SY, Gwi E, Ng HK, Fung CF, Yam KY. Dysembryoplastic neuroepithelial tumor. A tumor with small neuronal cells resembling oligodendroglioma. Am J Surg Pathol. 1994;18(6):604-614.
pubmed doi - Barth H, Fritsch G, Haaks T. [Intracerebral hematoma as an acute manifestation of intracranial tumors]. Nervenarzt. 1994;65(12):854-858.
pubmed - Fernandez C, Girard N, Paz Paredes A, Bouvier-Labit C, Lena G, Figarella-Branger D. The usefulness of MR imaging in the diagnosis of dysembryoplastic neuroepithelial tumor in children: a study of 14 cases. AJNR Am J Neuroradiol. 2003;24(5):829-834.
pubmed - Koeller KK, Dillon WP. Dysembryoplastic neuroepithelial tumors: MR appearance. AJNR Am J Neuroradiol. 1992;13(5):1319-1325.
pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Medical Cases is published by Elmer Press Inc.